Henrys lov om adsorption. Langmuir adsorptionsisoterm ligning. Lyofile spredningssystemer. Klassificering og generelle karakteristika for pav. Termodynamik og micelliseringsmekanisme. Strukturen af overfladeaktive miceller i vandige og kulbrintemedier. solubilisering
I tilfælde af interaktion mellem to atomer:
U er interaktionsenergien;
U = U + U TRÆK.
 - Lennard-Jones ligning
, c, b, m = konst
- Lennard-Jones ligning
, c, b, m = konst
I tilfælde af interaktion af atomer med en fast overflade er det nødvendigt at opsummere alle interaktioner.
x er afstanden til overfladen
r - virkningsradius for tiltrækningskræfterne
dV - volumen
n er antallet af overflademolekyler
U ADS. er adsorptionsinteraktionsenergien



I tilfælde af adsorption øges tiltrækningen. Og i tilfælde af interaktion af den ikke-polære-ikke-polære type, er adsorptionen overvejende lokaliseret i fordybningerne.
elektrostatisk interaktion.
Polært adsorbent - ikke-polært adsorbat
Ikke-polær adsorbent - polært adsorbat
En polær adsorbent er et polært adsorbat.
M  adsorbatmolekylet er repræsenteret som en dipol, og adsorbenten som en leder, hvor adsorbatmolekylet inducerer en dipol spejlsymmetrisk i forhold til den givne.
adsorbatmolekylet er repræsenteret som en dipol, og adsorbenten som en leder, hvor adsorbatmolekylet inducerer en dipol spejlsymmetrisk i forhold til den givne.
X - afstand til midten
Når du interagerer, opstår potentialet:
 ,
,
 er dipolmomentet.
er dipolmomentet.
Potentialet har en tendens til at få den maksimale værdi, dvs. dipoler har en tendens til at orientere sig vinkelret på overfladen.
Da en temperaturstigning fremmer væksten af Brownsk bevægelse, fører det til en deceleration af adsorptionsprocessen.
I tilfælde af elektrostatisk interaktion er adsorbatet overvejende lokaliseret på fremspring.
Fundamental adsorptionsligning.
Ved adsorption omfordeles komponenten, hvilket betyder, at det kemiske potentiale ændres. Adsorptionsprocessen kan betragtes som overgangen af overfladeenergi til kemisk energi.
Lagvolumen = 0, derefter den generaliserede ligning I og II for termodynamikkens lov:
T = const; (1) = (2) => 

For et to-komponent system:
,
 ,
,
 =>
=>
=>
 - Gibbs adsorptionsligning
.
- Gibbs adsorptionsligning
.
I tilfælde af adsorption af TV. krop - gas:, 
 ,
,

 - isoterm
- isoterm
 - isobar
- isobar
 - isopykne
- isopykne
 - isostere
- isostere
Isoterm, isopycne, isostere er relateret til hinanden.
Fordi adsorptionsfunktion 



Henry isoterm Langmuir isoterm



Termodynamik. Adsorption.
For kondenserede medier:
 ,
,
 ,
,
 - integreret ændring i Gibbs-energien
.
- integreret ændring i Gibbs-energien
.

P-tryk over en buet overflade, P S-tryk over en flad overflade
 - adsorptionspotentiale
- adsorptionspotentiale
Differentiel ændring i entrapi 
 , Г = konst
, Г = konst
- differentiel entropiændring
- differentiel entalpi af adsorption
 - isosterisk adsorptionsvarme
- isosterisk adsorptionsvarme
 - kondensationsvarme
- kondensationsvarme
 - netto adsorptionsvarme
- netto adsorptionsvarme

 ,
,




Qa er den integrerede adsorptionsvarme, 
Qra er den integrerede netto adsorptionsvarme,

Henrys ligning
Studiet af adsorption er hæmmet af overfladens inhomogenitet, så de enkleste regelmæssigheder opnås for homogene overflader.
Lad os overveje samspillet mellem gasser og en fast overflade, når en gas går fra en ligevægtstilstand i volumen til en ligevægtstilstand på overfladen. Dette tilfælde er analogt med ligevægten af gasser i et gravitationsfelt.
 ,
,
 ,
=>
,
=> -Henrys ligning
-Henrys ligning
 - fordelingskoefficient
- fordelingskoefficient
I adsorptionsprocessen sker der en ændring i kemiske potentialer.
For bulkfase: 
Til overfladegas: 
I en tilstand af ligevægt  , dvs.
, dvs.

I Henry-ligningen afhænger konstanten ikke af koncentrationen
Henry-ligningen er gyldig i området med lave tryk og koncentrationer. Efterhånden som koncentrationen stiger, er 2 typer af afvigelser fra Henrys lov mulige:
1 - positive afvigelser, D falder, A falder
2 - negative afvigelser, D - stiger, A - stiger.
Typen af afvigelse bestemmes af overvægten af en eller anden type adsorbent-adsorbat-interaktion.
Med en stærk klæbende interaktion øges aktivitetskoefficienterne - en positiv afvigelse. Ved sammenhængende interaktioner observeres negative afvigelser.
monomolekylær adsorption.
Langmuir isoterm.
De enkleste regelmæssigheder blev opnået i Henrys teori. Langmuir foreslog en teori, ifølge hvilken adsorption betragtes som en kvasi-kemisk reaktion. Hvori:
Overfladen er energimæssigt ensartet.
Adsorption er lokaliseret, hvert adsorptionscenter interagerer med et adsorbatmolekyle.
Adsorbatmolekyler interagerer ikke med hinanden.
Adsorption er monolag.

 - overflade,
- overflade,  - adsorbat,
- adsorbat,  - adsorptionskompleks.
- adsorptionskompleks.
 , derefter koncentrationen af adsorptionssteder:
, derefter koncentrationen af adsorptionssteder:  ,
, - begrænsning af adsorption.
- begrænsning af adsorption.
 , derefter reaktionskonstanten:
, derefter reaktionskonstanten: 

 - Langmuirs ligning.
- Langmuirs ligning.
Adsorption versus koncentration
1 )
)
 ,
,
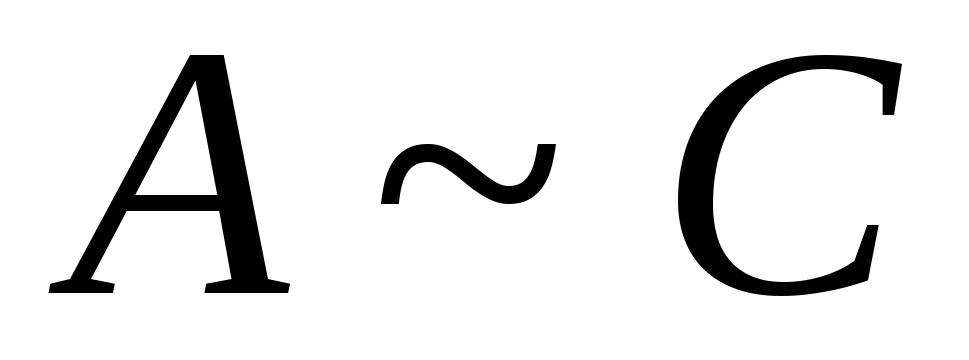
2) område med høje koncentrationer
 - begrænsning af adsorption, dannelse af et monomolekylært lag
- begrænsning af adsorption, dannelse af et monomolekylært lag
For Gibbs-energien: .
g er entropifaktoren.
I tilfælde af Henry-isotermen karakteriserer Gibbs-energien overgangen af adsorbatet fra standardtilstanden i bulken til standardtilstanden på overfladen. I tilfældet med Langmuir-isotermen  karakteriserer graden af affinitet af adsorbenten og adsorbatet.
karakteriserer graden af affinitet af adsorbenten og adsorbatet.
 fundet fra van't Hoff isobar.
fundet fra van't Hoff isobar.

 , derefter
, derefter  , derfor
, derfor  .
.

 - grad af overfladefyldning.
- grad af overfladefyldning.




 - antallet af ledige stillinger,
- antallet af ledige stillinger,  - antallet af besatte pladser.
- antallet af besatte pladser.
 ,
,

De der. i området med høje koncentrationer er antallet af frie steder omvendt proportionalt med mængden af adsorbat.
Adsorption af en blanding af gasser på en homogen overflade.
I dette tilfælde betragtes adsorptionsprocessen som to parallelle reaktioner.
 (1)
(1)

 (2)
(2)


Adsorption af en blanding af gasser på en inhomogen overflade.
Ved en ikke-homogen overflade bør man ikke begrænse sig til medium udfyldninger.
Som et resultat af konkurrence er lokalisering af forskellige adsorbater mulig på forskellige typer steder.
I dette tilfælde forholdet  .
.
 ,
,
 er adsorbatets mætningsdamptryk.
er adsorbatets mætningsdamptryk.
 ,
,
 er adsorptionsvarmen.
er adsorptionsvarmen.
"+" - symmatisk afhængighed, "-" - antibatisk afhængighed, "H" - ingen korrelation.
"+" - adsorption forløber efter samme mekanisme. I de mest energimæssigt gunstige områder adsorberes en gas med høj affinitet til overfladen overvejende.
"-" - adsorption forløber gennem forskellige mekanismer, og indtil et vist tidspunkt er der ingen konkurrence om overfladen.
Monomolekylær adsorption realiseres overvejende under den fysiske adsorption af gasser ved lave værdier s, samt ved væske/gas-grænsefladen.
Polymolekylær adsorption.
BET teori(Brunauer, Emmet, Teller).
I det tilfælde, hvor dannelsen af et monolag er utilstrækkelig til at kompensere for overfladeenergien, er adsorption polymolekylær og kan betragtes som et resultat af tvungen kondensation under påvirkning af overfladekræfter.
Grundlæggende bestemmelser:
Når et adsorbatmolekyle rammer det besatte sted, dannes et multipelt sæt.
Når du kommer tættere på s til s s antallet af gratis adsorptionssteder falder. I første omgang stiger antallet af pladser besat af singler, doubler osv. og falder derefter. sæt.
På s =s s adsorption bliver til kondens.
Der er ingen horisontale interaktioner.
For det første lag udføres Langmuir-isotermen.
Overfladen betragtes som et sæt adsorptionssteder. Betingelsen for dynamisk ligevægt er gyldig: kondensationshastigheden på frie steder er lig med fordampningshastigheden fra besatte.
a er kondensationskoefficienten (fraktionen af molekyler kondenseret på overfladen);
 ,
,
Zm er det maksimale antal ledige pladser.
 - hyppigheden af atomers svingninger i retningen vinkelret på overfladen.
- hyppigheden af atomers svingninger i retningen vinkelret på overfladen.
For det første lag er de dynamiske ligevægtsbetingelser:

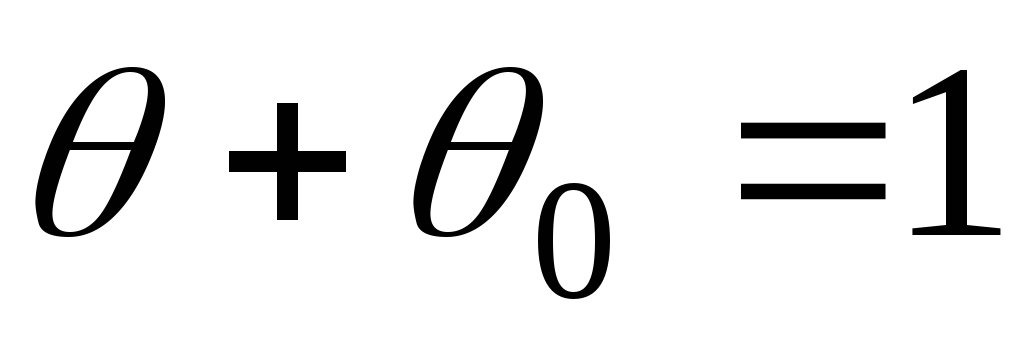

 , derefter
, derefter
 - Langmuirs ligning.
- Langmuirs ligning.
For det andet lag vil være sandt: 
For det i-te lag: 
For nemheds skyld antages det, at a og ν er ens for alle lag undtagen det første. For alle lag undtagen det første er adsorptionsvarmen konstant. For det sidste lag er adsorptionsvarmen lig med kondensationsvarmen. Som et resultat, ligningen
 (*)
(*)
C- konstant,
I tilfælde af BET-teori er konstanten FRA karakteriserer Gibbs energi af ren adsorption. Ligningen indeholder kun én konstant, og denne ligning er også meget vigtig for at bestemme det specifikke overfladeareal af adsorbenten.
Da varme frigives som følge af adsorption, udføres bestemmelsen af specifikke overflader ved lave temperaturer.
 ????????????
????????????


Teoriens største fejl– Forsømmelse af horisontale interaktioner til fordel for vertikale.
Ligningen er i området  fra 0,05 til 0,3.
fra 0,05 til 0,3.
Hvor  <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 > 0,3 - interaktionen af adsorbatet - adsorbat påvirker.
> 0,3 - interaktionen af adsorbatet - adsorbat påvirker.
Regnskab for adsorbat-adsorbat interaktioner.
Interaktioner forekommer under adsorption på en ikke-polær overflade af forgrenede molekyler eller molekyler. i stand til at danne foreninger. I dette tilfælde ændres formen af adsorptionsisotermerne.
MEN  sorbenten er ikke polær.
sorbenten er ikke polær.
Graf 1 svarer til svage interaktioner adsorbat-adsorbat, stærk adsorbat-adsorbent.
Graf 2 svarer til en stærk adsorbat-adsorbat-vekselvirkning, en stærk adsorbat-adsorbent-vekselvirkning.
Graf 3 svarer til en stærk adsorbat-adsorbat-interaktion, en svag adsorbat-adsorbent-vekselvirkning.
 ,
,

I tilfælde af interaktion mellem adsorbatmolekyler er det nødvendigt at tage højde for ændringer i aktivitetskoefficienterne. Og denne ligning er skrevet som:
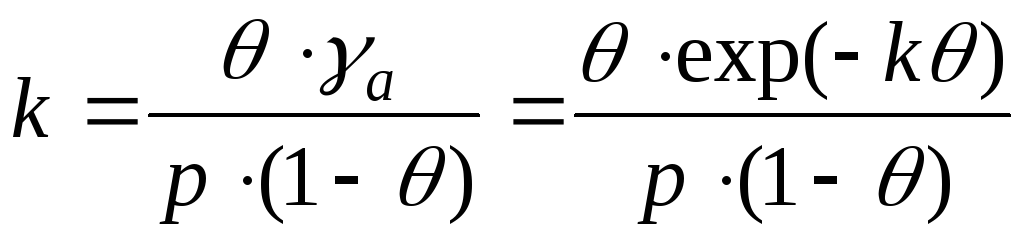 - ligningen af Frunkin, Fowler, Guggenheim.
- ligningen af Frunkin, Fowler, Guggenheim.
k er tiltrækningskonstanten.
Polans potentielle teori.
Denne teori udleder ikke nogen form for adsorptionsisoterm, men gør det muligt at beregne isotermer ved en anden temperatur.
Adsorption er resultatet af tiltrækningen af adsorbatet til overfladen af adsorbenten på grund af adsorptionspotentialets virkning, som ikke afhænger af tilstedeværelsen af andre molekyler og afhænger af afstanden mellem overfladen og adsorbatmolekylet.
 ,
,
 - adsorptionspotentiale.
- adsorptionspotentiale.
Da overfladen er inhomogen, erstattes afstanden af adsorptionsvolumenet  .adsorptionsvolumen er volumenet indesluttet mellem overfladen og punktet svarende til den givne værdi
.adsorptionsvolumen er volumenet indesluttet mellem overfladen og punktet svarende til den givne værdi  .
.
Adsorptionspotentiale er arbejdet med at overføre 1 mol af adsorbatet uden for det givne adsorptionsvolumen til et givet punkt af adsorptionsvolumenet (eller arbejdet med at overføre 1 mol mættet damp af adsorbatet, som er i ligevægt med det flydende adsorbat i fravær af adsorbenten, ind i dampfasen i ligevægt med adsorbenten).

Karakteristisk kurve
 - adsorptionspotentiale,
- adsorptionspotentiale, 
For en given adsorbent og forskellige adsorbater gælder følgende: 
Til forskellige typer adsorbater  ,
,
hvor  potentialer for adsorptionsisotermer ved relative tryk
potentialer for adsorptionsisotermer ved relative tryk  for adsorbat 1 og for adsorbat 2. Dette forhold er en konstant værdi.
for adsorbat 1 og for adsorbat 2. Dette forhold er en konstant værdi.
 - affinitetskoefficient
- affinitetskoefficient
Teori om kapillær kondensation.
Forløbet af adsorptionsprocessen afhænger i høj grad af strukturen af det porøse legeme.
|
mikroporøs | |
|
Overgangsporøs | |
|
Makroporøst |
I tilfælde af mikroporøse sorbenter overlapper felterne af adsorptionskræfter hinanden. Ved makroporøse sorbenter fungerer porerne som transportkanaler. Kondensationsprocesserne er mest betydningsfulde i forbigående porøse legemer. Kapillærkondensering starter ved bestemte værdier s Og  når en del af overfladeenergien allerede er kompenseret. En nødvendig betingelse er, at overfladen skal være selvbærende. Processen er beskrevet Thompson-Kelvin ligning.
når en del af overfladeenergien allerede er kompenseret. En nødvendig betingelse er, at overfladen skal være selvbærende. Processen er beskrevet Thompson-Kelvin ligning.

 - i tilfælde af befugtning er krumningscentret i gasfasen.
- i tilfælde af befugtning er krumningscentret i gasfasen.
I tilfælde af kapillær kondensation har adsorptionsisotermen en hystereseform. Den nederste gren svarer til adsorptionsprocessen, og den øverste gren svarer til desorptionsprocessen.
Alle typer porer kan reduceres til tre typer:
|
konisk |
Cylindrisk med en lukket ende |
Cylindrisk med to åbne ender |
|
Procesfyldning udføres fra bunden af poren. Adsorptionsisotermen og desorptionsisotermen falder i dette tilfælde sammen, da adsorptionsprocessen begynder med en kugle, og desorptionsprocessen begynder også med forsvinden af nogle kugler.
↓ |
Der er ingen hysterese. Frem- og tilbageslag beskrives ved ligningen:
|
Der er ingen bund nogen steder, fyldningen af poren vil gå langs cylinderens vægge.
cylinder: Isoterm og vil have en hystereseform.
↓ |


 - sfære,
- sfære, ,
,



I  befugtningsforhold opstår der kondens ved lavere tryk, hvilket er energetisk gunstigt. Fra desorptionsgrenen opnås porestørrelsesfordelingskurver.
befugtningsforhold opstår der kondens ved lavere tryk, hvilket er energetisk gunstigt. Fra desorptionsgrenen opnås porestørrelsesfordelingskurver.
Differentialkurvens maksimum forskydes til venstre i forhold til integralets bøjningspunkt. Det samlede volumen af små porer er lille, men har store overfladearealer. Når porestørrelsen øges, øges deres volumen som  , og området som
, og området som  På grund af dette observeres en forskydning af differentialkurvens maksimum.
På grund af dette observeres en forskydning af differentialkurvens maksimum.
Adsorption ved fast-væske-grænsefladen.
I tilfælde af adsorption ved fast-gas-grænsefladen forsømte vi en komponent. I tilfælde af adsorption ved faststof-væske-grænsefladen fortrænger adsorbatet opløsningsmiddelmolekyler fra overfladen af adsorbenten.
 ,
,
Den rigtige ligning er:

 ,
,
N 1, N 2 - molfraktioner af opløsningsmidlet og komponenten, N 1 + N 2 \u003d 1, derefter
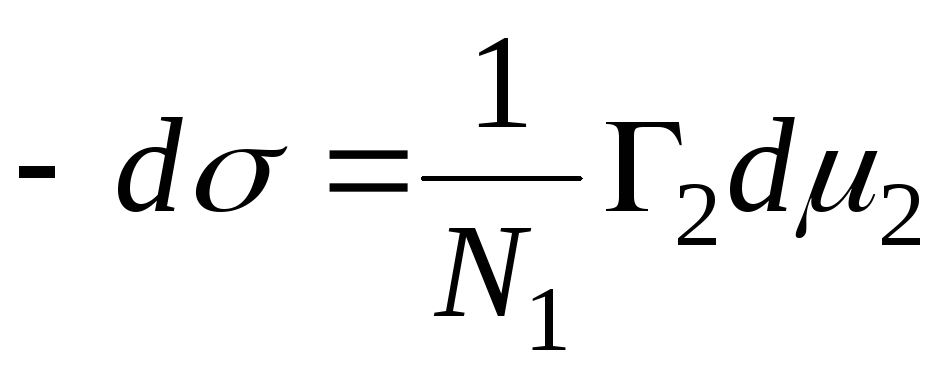 ,
=>
,
=>
 , derefter - adsorptionsligningen for fasegrænsen faststof - væske.
, derefter - adsorptionsligningen for fasegrænsen faststof - væske.
Adsorption (G) > 0 ved  <
0
<
0
Hvis værdierne  for komponenten og opløsningsmidlet er meget forskellige, i dette tilfælde afhængigheden G fra N har et ekstremum ved værdien N
~ 0,5.
for komponenten og opløsningsmidlet er meget forskellige, i dette tilfælde afhængigheden G fra N har et ekstremum ved værdien N
~ 0,5.
E  hvis
hvis  har lignende værdier, i dette tilfælde kan tegnet på adsorption ændre sig. Afhængighed G fra N krydser x-aksen
har lignende værdier, i dette tilfælde kan tegnet på adsorption ændre sig. Afhængighed G fra N krydser x-aksen
Funktion Skæring G(N) med abscisseaksen kaldes adsorptionsazeotrop. Det betyder, at de to komponenter ikke kan adskilles på denne adsorbent.
Adsorptionsisotermligning med udvekslingskonstant.
Under adsorption ved faststof-væske-grænsefladen omfordeles komponenterne konstant mellem overfladen af adsorbenten og volumenet af opløsningen.
 - komponenter (- - referer til overfladen)
- komponenter (- - referer til overfladen)

 ,
,
 ,
, .
.
 ,
,


Adsorption ved væske-gas-grænsefladen
R  Lad os overveje ændringen i koncentrationsprofilen, når væske-gas-grænsefladen krydses. Lad komponent 2 være flygtig.
Lad os overveje ændringen i koncentrationsprofilen, når væske-gas-grænsefladen krydses. Lad komponent 2 være flygtig.
Cs er koncentrationen i overfladelaget.
Baseret på definitionen af overskydende adsorption

Hvis komponenten ikke er flygtig, vil adsorptionsværdien blive skrevet som følger:
P  ri
ri 

I ligningen  materiens natur er beskrevet af derivatet
materiens natur er beskrevet af derivatet  .
.
Overfladespændingsisotermen kan have formen 1 eller 2:
1 - overfladeaktive stoffer
2 - overfladeaktive stoffer
Overfladeaktivitet g er stoffers evne til at reducere overfladespændingen i systemet.

 - tykkelse af overfladelaget
- tykkelse af overfladelaget
C s er koncentrationen af komponenten i overfladelaget
FRA– volumenkoncentration
For en homolog serie er der en regel:
 - Traubeau Duclos-reglen
- Traubeau Duclos-reglen
For den homologe serie ser adsorptionsisotermen sådan ud:
Vi skriver D i stedet for A, da adsorptionen er for høj i overfladelaget.
Overfladespændingsisoterm:

 er overfladespændingen af det rene opløsningsmiddel.
er overfladespændingen af det rene opløsningsmiddel.
 - fundamental adsorptionsligning;
- fundamental adsorptionsligning;
 - Langmuirs ligning.
- Langmuirs ligning.
Lad os løse dem sammen:

- Shishkovskys ligning.
B er en konstant for den homologe serie.
EN- når man flytter fra en homolog til en anden, stiger den med 3-3,5 gange 
![]()
1 - område med lave koncentrationer
![]()
2 - gennemsnitlig koncentration
3 - monomolekylært lag
Overfladeaktive stoffer er amfifile molekyler, dvs. omfatter en polær gruppe og et ikke-polært carbonhydridradikal.
o er den polære del af molekylet.
| er den ikke-polære del af molekylet.
I et polært opløsningsmiddel er overfladeaktive molekyler orienteret på en sådan måde, at den polære del af molekylet vender mod opløsningsmidlet, mens den upolære del skubbes ind i gasfasen.
I Shishkovsky-ligningen  , den er konstant for den homologe serie.
, den er konstant for den homologe serie.
Overfladeaktiv handling begynder at dukke op med n>5. Ved koncentrationer højere end koncentrationen af det monomolekylære lag forekommer micellisering i overfladeaktive opløsninger.
Micelle- kaldes aggregatet af amfifile overfladeaktive molekyler, hvis carbonhydridradikaler danner kernen, og de polære grupper omdannes til den vandige fase.
Micellemasse - micellær masse.
H  antallet af molekyler er antallet af aggregering.
antallet af molekyler er antallet af aggregering.
Kugleformede miceller
Ved micellisering etableres en ligevægt i opløsningen
CMC er den kritiske micellekoncentration.
Da vi betragter micellen som en separat fase:
For den homologiske serie er der en empirisk ligning:
-en er den funktionelle gruppes opløsningsenergi.
b er stigningen i adsorptionspotentialet, arbejdet med adsorption pr. methylenenhed.
er stigningen i adsorptionspotentialet, arbejdet med adsorption pr. methylenenhed.
Tilstedeværelsen af en kulbrintekerne i miceller gør det muligt for forbindelser, der er uopløselige i vand, at opløses i vandige opløsninger af overfladeaktive stoffer, dette fænomen kaldes solubilisering (det der opløses er et solubilisat, overfladeaktivt stof er et solubiliseringsmiddel).
Mudderet kan være fuldstændig upolært, kan indeholde både polære og ikke-polære dele og vil være orienteret som et overfladeaktivt molekyle.
Under alle omstændigheder sker der under solubilisering en stigning i micellarmassen og aggregeringstallet ikke kun på grund af inklusion af solubilisatet, men også på grund af en stigning i antallet af overfladeaktive molekyler, der er nødvendige for at opretholde ligevægtstilstanden.
Solubilisering er jo mere effektiv, jo lavere molekylvægten af solubilisatet er.

 ~ 72 mN/m.
~ 72 mN/m.
 ~ 33 mN/m.
~ 33 mN/m.
Effektiviteten af overfladeaktive stoffer afhænger af størrelsen af CMC.
2D overfladelagstryk

→ -kræfter af overfladespænding.
- todimensionelt tryk.
Overfladelaget er en kraft svarende til forskellen mellem overfladespændingerne af en overfladeaktivt opløsning og et rent opløsningsmiddel, rettet mod en ren overflade.
Der etableres en ligevægt mellem opløsningen og overfladelaget



På  der er et område hvor
der er et område hvor  lineært afhængig af koncentration.
lineært afhængig af koncentration.
G [mol/m2].
 område optaget af et mol af et stof
område optaget af et mol af et stof
Så vil den todimensionelle trykisoterm have formen 
 er den todimensionelle trykisoterm.
er den todimensionelle trykisoterm.
Afhængighed  fra S M:
fra S M:

På  - det todimensionelle tryk stiger kraftigt. På
- det todimensionelle tryk stiger kraftigt. På  todimensional er deformeret, hvilket forårsager en kraftig vækst
todimensional er deformeret, hvilket forårsager en kraftig vækst  .
.
Filmen på begge sider begrænset af de samme faser kaldes dobbeltsidet. I sådanne film observeres en konstant bevægelse af moderluden.
Film med en tykkelse på mindre end 5 nm kaldes sorte film.

Adsorptionslag bør have to egenskaber: viskositet og let mobilitet, flydende og elasticitet.
Marangoni-effekten er selvhelbredende.
Gibbs trekant,  - overtryk.
- overtryk.
Filmen strækkes, og på grund af det faktum, at en del af væsken er væk, skynder de overfladeaktive stoffer ind i det frie rum. Gibbs trekant.
Effekt af legemers adsorptionsstyrke.
Der er altid et adsorptionslag på filmoverfladen, for hvilket , så
Langmuir ligning:


 ind i todimensionalt tryk
ind i todimensionalt tryk
 - analog af Shishkovsky-ligningen
- analog af Shishkovsky-ligningen
elektrokinetiske fænomener. Dobbelt elektrisk lag (DES).
Helemholtz model. Gouy-Chapman teori.
1808 Flyvning

U – formet rør, nedsænket i det 2 elektroder. Loven om kommunikerende fartøjer er overtrådt, og der er en ændring i væskeniveauet i røret - elektrokinetiske fænomener.
Kinetiske fænomener:
elektroforese
Elektroosmose
Flow (flow) potentiale
Sedimentationspotentiale
1 og 2 opstår, når en potentialforskel påføres, 3 og 4 forårsager udstansningen og sedimenteringen af kolloide partikler fremkomsten af en potentialforskel.
Elektroosmose er bevægelsen af et dispersionsmedium i forhold til en stationær spredt fase under påvirkning af en elektrisk strøm.
elektroforese er bevægelsen af partikler af den dispergerede fase i forhold til et stationært dispersionsmedium under påvirkning af en elektrisk strøm.
P  Årsagen til forekomsten af elektrokinetiske fænomener er den rumlige adskillelse af ladninger og udseendet af et dobbelt elektrisk lag.
Årsagen til forekomsten af elektrokinetiske fænomener er den rumlige adskillelse af ladninger og udseendet af et dobbelt elektrisk lag.
Det elektriske dobbeltlag er en flad kondensator, den ene plade er dannet af potentialbestemmende ioner, den anden af kontrainoer. Ionerne er også forurenet, da potentialbestemmende co-ioner skubbes ind i hovedparten af opløsningen. Afstand mellem plader  . Potentialet falder lineært, potentialforskellen
. Potentialet falder lineært, potentialforskellen  .
.
En ekstern potentialforskel forårsager fremkomsten af et forskydningsmodul  er et par kræfter pr. arealenhed, der virker langs overfladen af et fast legeme.
er et par kræfter pr. arealenhed, der virker langs overfladen af et fast legeme.

Ved ligevægt er forskydningsmodulet lig med det viskøse friktionsmodul (  ).
).

I vores forhold  ,
,

 - Helemholtz-Smalukovsky ligning
- Helemholtz-Smalukovsky ligning
 - lineær hastighedsforskydning i faser.
- lineær hastighedsforskydning i faser.
E er den elektriske feltstyrke.
 - potentialforskel mellem pladerne
- potentialforskel mellem pladerne
 - elektroforetisk mobilitet [m 2 /(V * s)].
- elektroforetisk mobilitet [m 2 /(V * s)].
Helemholtz-modellen tager ikke højde for molekylers termiske bevægelse. I virkeligheden er fordelingen af ioner i dobbeltlaget mere kompleks.
Gouy og Chapman identificerede følgende årsager til DES:
Overgangen af en ion fra en fase til en anden, når ligevægt er etableret.
Ionisering af fastfasestof.
Fuldførelse af overfladen med ioner til stede i dispersionsmediet.
Polarisering fra en ekstern strømkilde.
Det elektriske dobbeltlag har en sløret eller diffus struktur. Ioner har en tendens til at være jævnt fordelt gennem det diffuse lag.
Det diffuse lag består af modinioner, lagets længde bestemmes af deres kinetiske energi. Ved en temperatur, der har tendens til det absolutte nulpunkt, er kontrainoer så tæt som muligt på potentialebestemmende ioner.
Denne teori er baseret på to ligninger:
Boltzmann ligning

 - arbejde mod kræfterne fra elektrostatisk interaktion.
- arbejde mod kræfterne fra elektrostatisk interaktion.
 er bulk ladningstætheden.
er bulk ladningstætheden.

Poisson ligning 
Da DEL-tykkelsen er meget mindre end partikelstørrelsen, og for en flad DEL, er den afledte med hensyn til koordinater  Og
Og  er afskaffet.
er afskaffet. 
For e y med y<<1 функцию можно разложить в ряд Маклорена:

Vi begrænser os til to medlemmer af serien, så:


 - DES tykkelse er den afstand, hvormed DES potentialet falder i e enkelt gang.
- DES tykkelse er den afstand, hvormed DES potentialet falder i e enkelt gang.
Jo lavere temperatur, jo mindre  . Ved Т→0 – flad DES. Jo højere koncentration, jo mere I, jo mindre
. Ved Т→0 – flad DES. Jo højere koncentration, jo mere I, jo mindre  .
.





 "–" betyder, at potentialet falder med afstanden. =>
"–" betyder, at potentialet falder med afstanden. =>
=>

 ,
,
 - potentialet falder eksponentielt.
- potentialet falder eksponentielt.
Potentiale for overfladeladningstæthed: 
Overfladeladning er en rumladning med det modsatte fortegn, integreret over afstand.



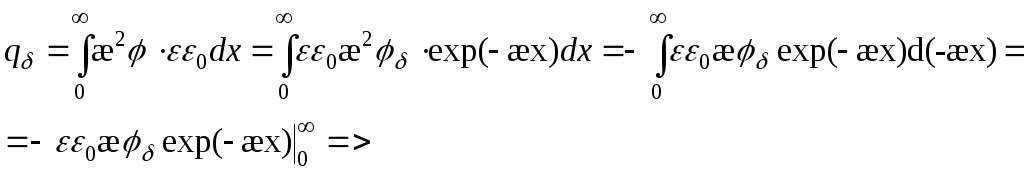
=>


Hvor potentialet falder med 2,7 gange - 
Dobbelt lag kapacitet 
Ulempen ved teorien er, at tilstedeværelsen af Helemholtz-laget ikke tages i betragtning, dvs. tager ikke hensyn til  , deraf fejlene ved bestemmelse af hovedparametrene. Det forklarer heller ikke virkningen af ioner af forskellig natur på tykkelsen af det elektriske dobbeltlag.
, deraf fejlene ved bestemmelse af hovedparametrene. Det forklarer heller ikke virkningen af ioner af forskellig natur på tykkelsen af det elektriske dobbeltlag.
Sterns teori. Strukturen af en kolloid micelle.
Det elektriske dobbeltlag består af to dele: tæt og diffust. Et tæt lag dannes som et resultat af interaktionen af potentialdannende ioner med specifikt adsorberede. Disse ioner er som regel delvist eller fuldstændigt dehydrerede og kan have enten den samme eller den modsatte ladning til de potentialbestemmende ioner. Det afhænger af forholdet mellem energien af elektrostatisk interaktion  og specifikt adsorptionspotentiale
og specifikt adsorptionspotentiale  . Ionerne i det tætte lag er fikseret. Den anden del af ionerne er placeret i det diffuse lag, disse ioner er frie og kan bevæge sig dybt ind i opløsningen, dvs. fra et område med højere koncentration til et område med lavere koncentration. Den samlede ladningstæthed består af to dele.
. Ionerne i det tætte lag er fikseret. Den anden del af ionerne er placeret i det diffuse lag, disse ioner er frie og kan bevæge sig dybt ind i opløsningen, dvs. fra et område med højere koncentration til et område med lavere koncentration. Den samlede ladningstæthed består af to dele.

 - Helmholtz lag ladning
- Helmholtz lag ladning
 -Diffus lagladning
-Diffus lagladning
Overfladen har et vist antal adsorptionscentre, som hver interagerer med en modion. Konstanten for en sådan kvasikemisk reaktion er:

 , hvor
, hvor  - molfraktion af modioner i opløsning
- molfraktion af modioner i opløsning
Helmholtz distribution
Potentialet falder lineært
Gouy-potentialefordeling. Der er ikke noget tæt lag, potentialet falder eksponentielt fra værdien 
Stern fordeling.
Til at begynde med er det potentielle fald lineært og derefter eksponentielt.
Når et elektrisk felt påføres i tilfælde af elektroforese, er det ikke partiklen af den faste fase, der bevæger sig direkte, men partiklen af den faste fase med et lag af omgivende ioner. DES gentager formen af partiklen i den dispergerede fase. Når et potentiale påføres, rives en del af det diffuse lag af. Brydelinjen kaldes glidende grænse.
Det potentiale, der opstår ved glidegrænsen som følge af løsrivelse af en del af det diffuse lag, kaldes elektrokinetisk potentiale(Zeta potentiale  ).
).
En partikel af en dispergeret fase, med et lag af modioner, der omgiver den og et dobbelt elektrisk lag, kaldes micelle.
Regler for skrivning af kolloide miceller:


1-1 ladeelektrolyt
T er en partikel af den dispergerede fase.
AA er grænsen mellem de tætte og diffuse dele.
BB er glidegrænsen.
Slipgrænsen falder muligvis ikke sammen med linje AA.
Den pH-værdi, ved hvilken zeta-potentialet er nul, kaldes isoelektrisk punkt.
CaCl 2 + Na 2 SO 4 → CaSO 4 ↓ + 2 NaCl
1. Over CaCl 2
CaCl 2 ↔ Ca 2+ + 2Cl -
(CaSO 4 m∙nCa 2+ 2( n-x)Cl-) 2 x + x Cl - - optage miceller.
CaSO 4 m - aggregat.
CaSO 4 m∙nCa 2+ er kernen.
CaSO 4 m∙nCa 2+ 2( n-x)Cl - partikel.
2. I overskud af Na 2 SO 4
Na 2 SO 4 ↔2Na + + SO 4 2-
(CaSO 4 m∙nSO 4 2- 2(n-x)Na+) 2x- 2xNa+ - micelle
CaSO 4 m - aggregat.
CaSO 4 m∙nSO 4 2 + er kernen.
CaS04 m∙nSO4 2-2(n-x)Na+-partikel
Helemholtz-Smoluchowski ligning

 - lineær hastighed for forskydning af grænser (ved elektroosmose).
- lineær hastighed for forskydning af grænser (ved elektroosmose).
 - potentialforskel på kondensatorpladerne (ved elektroosmose).
- potentialforskel på kondensatorpladerne (ved elektroosmose).


 - volumetrisk strømningshastighed af opløsningen, S er cellens tværsnitsareal.
- volumetrisk strømningshastighed af opløsningen, S er cellens tværsnitsareal.

E er den elektriske feltstyrke.


 (til elektroosmose).
(til elektroosmose).
For flowpotentialet:

 - potentiale
- potentiale
 - membrantryk
- membrantryk
Som regel er værdien af elektroforetiske mobiliteter og elektroosmotiske mobiliteter mindre end de beregnede. Dette skyldes:
Afslapningseffekt (under bevægelsen af en partikel af den dispergerede fase krænkes symmetrien af den ioniske atmosfære).
Elektroforetisk bremsning (forekomsten af yderligere friktion som følge af bevægelsen af modioner).
Forvrængning af strømlinjer i tilfælde af elektrisk ledende partikler.
Sammenhæng mellem overfladespænding og potentiale. Lippmann ligning.
Dannelsen af DEL sker spontant på grund af systemets ønske om at reducere dets overfladeenergi. I sammenhæng med konstanthed T Og s den generaliserede ligning af termodynamikkens første og anden lov ser sådan ud:
 (2)
(2)
(3), (1)=(3) =>
=>

 - 1. Lippmann-ligning.
- 1. Lippmann-ligning.
 er overfladeladningstætheden.
er overfladeladningstætheden.
 - differentiel kapacitans.
- differentiel kapacitans.
 - 2. Lippmann-ligning.
- 2. Lippmann-ligning.
FRA- kapacitet.
Vi løser den 1. Lippmann-ligning og den fundamentale adsorptionsligning:
 ,
,


 , derefter
, derefter
 - Nernst-ligningen
- Nernst-ligningen
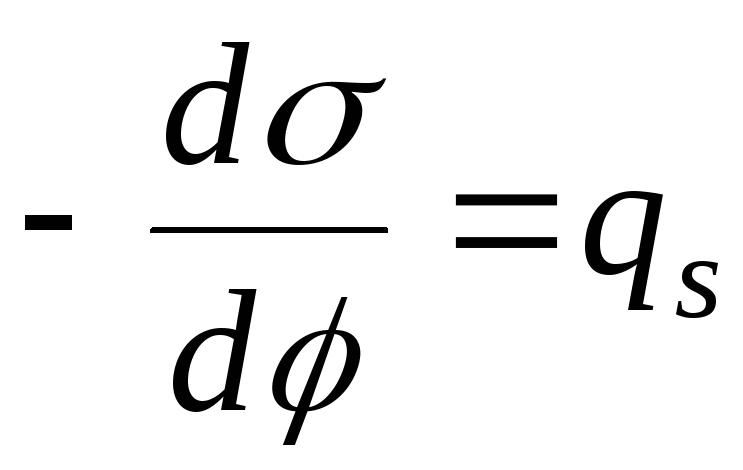 ,
,
 ,
,




 - ligningen for den elektrokapillære kurve (ECC).
- ligningen for den elektrokapillære kurve (ECC).
I  :
: , men
, men 
Kationiske overfladeaktive stoffer (CSAS) reducerer den katodiske gren af ECC.
Anioniske overfladeaktive stoffer (ASS) reducerer den anodiske gren af ECC.
Non-ioniske overfladeaktive stoffer (NSA) reducerer den midterste del af ECC.
Stabilitet af disperse systemer. Kiletryk.
Dispergerede systemer kan opdeles:
Termodynamisk ustabile systemer kan være kinetisk stabile på grund af overgangen til en metastabil tilstand.
Der er to typer stabilitet:
Sedimentationsstabilitet (med hensyn til tyngdekraften).
Aggregativ stabilitet. (i forhold til at stikke)
Koagulering er processen med at partikler klæber sammen, hvilket fører til tab af aggregatstabilitet. Koagulation kan være forårsaget af ændringer i temperatur, pH, omrøring, ultralyd.
Skelne koagulation:
Vendbar.
Irreversibel.
Koagulation fortsætter med indførelse af elektrolytter.
Koagulationsregler:

Film- Dette er den del af systemet, der er placeret mellem to grænseflader.
usammenhængende pres opstår med et kraftigt fald i filmtykkelsen som følge af vekselvirkningen mellem nærliggende overfladelag.

«-» - når filmtykkelsen falder, stiger det adskillende tryk.
P 0 er trykket i bulkfasen, som er en fortsættelse af mellemlaget.
P 1 er trykket i filmen.
Teori om stabilitet. DLFO (Deryagin, Landau, Fairway, Overbeck).
Ifølge DLVO-teorien skelnes der mellem to komponenter i det usammenhængende tryk:
elektrostatisk PE (positiv, det skyldes kræfterne fra elektrostatisk frastødning). Svarer til et fald i Gibbs-energien med stigende filmtykkelse.
Molekylær PM (negativ, på grund af virkningen af tiltrækkende kræfter). Det er forårsaget af komprimeringen af filmen på grund af kemiske overfladekræfter, kræfternes virkningsradius er tiendedele af en nm med en energi i størrelsesordenen 400 kJ/mol.
Total interaktionsenergi:

 - systemet er samlet stabilt
- systemet er samlet stabilt
 - ustabilt system
- ustabilt system
P  positiv komponent.
positiv komponent.
Stigningen skyldes stigningen i potentiel energi under kompression af tynde film. For tykke film kompenseres den overskydende ionenergi og er lig med energiinteraktionen i hovedparten af dispersionsmediet.
Hvis  (
( - filmtykkelse,
- filmtykkelse,  - ionens radius) udtynding af filmen fører til forsvinden og reduktion i den af molekyler og ioner med en minimal overfladeenergi. Antallet af nabopartikler falder, som et resultat af hvilket den potentielle energi af de partikler, der er tilbage i filmen, stiger.
- ionens radius) udtynding af filmen fører til forsvinden og reduktion i den af molekyler og ioner med en minimal overfladeenergi. Antallet af nabopartikler falder, som et resultat af hvilket den potentielle energi af de partikler, der er tilbage i filmen, stiger.


DLVO-teorien betragter interaktionen af partikler som interaktionen af plader.

Partikler interagerer ikke



 - Laplace ligning,
- Laplace ligning,  ,
,


Til svagt ladede overflader
Til stærkt ladede overflader:

Den molekylære komponent er interaktionen mellem to atomer:
 ~
~
Interaktion mellem et atom og en overflade:

Lad os tage to optegnelser:
D  For at opnå den molekylære komponent er det nødvendigt at opsummere alle interaktionsenergierne for atomerne på højre og venstre plade.
For at opnå den molekylære komponent er det nødvendigt at opsummere alle interaktionsenergierne for atomerne på højre og venstre plade.
hvor  - Hamakers konstant (tager hensyn til arten af interagerende kroppe).
- Hamakers konstant (tager hensyn til arten af interagerende kroppe).
At. interaktionsenergien for partikler i et system kan udtrykkes ved hjælp af potentialkurver.
I er det primære potentielle minimum. Dette er en zone med irreversibel koagulation, tiltrækningskræfterne sejrer.
II - zone med aggregativ stabilitet, frastødende kræfter hersker.
III - sekundært potentiale minimum (eller flokkuleringszone). Mellem partiklerne i den dispergerede fase er der et elektrolytlag, og partiklerne kan adskilles og overføres til zonen med aggregativ stabilitet.

Kurve 1 – systemet er samlet set stabilt.
Kurve 2 er stabil i zone I, ikke stabil i zone II.
Kurve 3 - koagulering forekom i systemet.
Kurve 4 - ved punkt 4, den totale energi af interaktion U=0,  , svarer dette ekstremumpunkt til begyndelsen af hurtig koagulation.
, svarer dette ekstremumpunkt til begyndelsen af hurtig koagulation.
Der er to tilfælde:
1. Overflader er svagt ladede:
U \u003d U E + U M \u003d 0
 (1)
(1)
2)

 (2)
(2)



 - dette er tykkelsen af laget svarende til begyndelsen af koaguleringsprocessen.
- dette er tykkelsen af laget svarende til begyndelsen af koaguleringsprocessen.






 - til svagt ladede overflader
- til svagt ladede overflader
 derefter
derefter 
2. For stærkt ladede overflader:
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
Lad os firkante (3)

Koagulering:
Ved specifik adsorption kan ioner adsorberes i en superækvivalent mængde på en sådan måde, at overfladen kan ændre sin ladning. Overfladen genoplades.
I tilfælde af specifik adsorption kan ikke kun ioner med modsatte fortegn, men også af en ion adsorberes.
Hvis ioner af samme fortegn som overfladen adsorberes, så vil der i overfladelaget ikke være et fald i potentialet, men dets vækst.

Neutraliseringskoagulering (opstår med deltagelse af svagt ladede partikler og afhænger ikke kun af ladningen af den koagulerende elektrolyt, men også af potentialet ved grænsen af de tætte og diffuse lag).
Smoluchowskis teori om hurtig koagulation.

Koagulationshastighedens afhængighed af elektrolytkoncentrationen.
I – koagulationshastigheden er lav,
II - koagulationshastigheden er praktisk talt proportional med elektrolytkoncentrationen.
III - området med hurtig koagulation, hastigheden er praktisk talt uafhængig af koncentration.
Centrale punkter:
Den oprindelige sol er monodispers, lignende partikler har en sfærisk form.
Alle partikelkollisioner er effektive.
Når to primære partikler kolliderer, dannes en sekundær partikel. Sekundær + primær = tertiær. Primær, sekundær, tertiær - multiplicitet.
Med hensyn til kemisk kinetik kan koaguleringsprocessen beskrives ved ligningen:

Løsningen bliver ligningen: 
 - tidspunkt for halvkoagulering. Dette er den tid, hvor antallet af solpartikler falder 2 gange.
- tidspunkt for halvkoagulering. Dette er den tid, hvor antallet af solpartikler falder 2 gange.

 ,
,
 ,
,
 ,
,


Efterhånden som multipliciteten øges, skifter maksimum af koagulationskurverne mod større værdier  .
.
Ulemper:
Antagelse om monodispersitet.
Antagelsen om effektiviteten af alle kollisioner.
Henry adsorption isoterm ligning
Hvis vi betragter det dynamiske billede af adsorption, så vil dets værdi være jo større, jo større antallet af påvirkninger af gasmolekyler på overfladen (dvs. jo større gastryk) og jo længere tid molekylet forbliver på overfladen fra stødmomentet til det øjeblik, det går tilbage til gasfasen. Derfor, men de Boer, adsorptionsværdien
hvor n er det gennemsnitlige antal molekyler, der rammer overfladen pr. tidsenhed, τ er den gennemsnitlige opholdstid for molekylerne på overfladen. Denne formel antager, at hvert påvirkning af et molekyle er ledsaget af dets forsinkelse på overfladen, uanset om der allerede er andre molekyler på det eller ej. I virkeligheden kan et molekyle, der rammer et allerede optaget sted, blive reflekteret tilbage i gasfasen eller forsinket, men dets retentionstid vil være anderledes.
Regnskab for disse omstændigheder førte til følgende formel:
Dette er Henrys adsorptionsisoterm-ligning. Det betyder, at i den ideelle model er mængden af adsorption direkte proportional med damp- eller gastrykket. Denne afhængighed modtog dette navn i analogi med Henrys lov kendt i fysisk kemi, ifølge hvilken volumenet af en gas opløst i et fast stof eller en væske er proportional med dets tryk. Så ifølge de accepterede antagelser skal Henry-isotermen beskrive de eksperimentelle data opnået ved lave fyldninger på homogene overflader. Den første antagelse er som sagt begrundet i undersøgelsen af adsorption ved meget lave tryk. Hvad angår det andet, måles adsorption næsten altid på inhomogene overflader. Adsorption ved meget lave tryk svarer dog til meget lave dækningsgrader. Det betyder, at alt afhænger af, hvor inhomogen ikke hele overfladen er, men kun en lille brøkdel af den, som dækkes ved lavtryk. Derfor kan man i litteraturen finde eksempler nok af begge slags. Konstanten K i Henry-ligningen (tangens til hældningen af den rette linje) afhænger af adsorbatets - adsorbentens temperatur og interaktionsenergi, som det kan ses af ligning (4.4). Jo lavere temperatur og jo større interaktion mellem adsorberede molekyler og overfladen af adsorbenten er, jo større K, jo stejlere er adsorptionsisotermen.
Selvfølgelig er antagelsen om, at molekyler adsorberes med samme sandsynlighed på en hvilken som helst del af overfladen, inklusive dem, der allerede er optaget tidligere, en for grov antagelse, der kun er egnet til meget små dækningsgrader. En anden antagelse kan gøres, som er, at adsorption kun sker på frie områder af overfladen, og at ethvert hit af molekyler på allerede besatte steder ikke fører til en adsorptionshændelse. Denne antagelse svarer til postulatet om monolagsadsorption, og som vi sagde tidligere, gælder den faktisk i tilfælde af kemisk adsorption, men situationen er mere kompliceret for fysisk adsorption.
En anden antagelse i udledningen af Henrys isotermligning er, at overfladen er homogen, dvs. ækvivalens af alle dens sektioner, vil vi holde uændret. Og endelig er den tredje antagelse i den nye model under overvejelse fraværet af interaktion mellem adsorberede molekyler, dvs. vi vil antage, at opholdstiden for et molekyle på overfladen ikke afhænger af, hvor det rammer - i umiddelbar nærhed af et andet molekyle eller i stor afstand fra det. Alle disse antagelser blev lavet af Langmuir i udledningen af adsorptionsisotermen, som han lavede i 1918.
Langmuirs adsorptionsisoterm-ligning kan udledes på forskellige måder. Langmuir selv udledte det ved at overveje afhængigheden af adsorptions- og desorptionshastighederne på graden af overfladedækning og antage, at begge hastigheder ved ligevægt bliver de samme.
Den termodynamiske udledning af denne ligning blev givet af Volmer, og den statistiske udledning af Fowler.
I denne form er Langmuir-ligningen almindeligt kendt. Den indeholder to konstanter: en m kort kaldet monolagskapacitet (maksimal adsorption), og K er en konstant afhængig af adsorptionsenergi og temperatur.
adsorptionsisoterm. Freundlich ligning.
Adsorptionsværdi (absolut MEN eller overskud G) i hvert tilfælde bestemmes af temperaturen T og tryk R(med et gasformigt adsorptionsmiddel) eller temperatur T og koncentration FRA(når adsorberet fra opløsninger). Som regel, i teorien om adsorption, når man overvejer adsorptionsligevægten, holdes en af disse parametre konstant. Så, en ligning på formen A \u003d f (p) T eller G \u003d f (c) T, der relaterer mængden af adsorption med tryk eller koncentration ved en konstant temperatur, kaldes adsorptionsisotermen. Adsorption (hvis det ikke udtrykkes som et overskud, men som et samlet indhold) stiger altid med stigende ligevægtstryk eller koncentration. Da adsorption er en eksoterm proces, når temperaturen stiger, værdien adsorption falder. På fig. 26.9 viser hovedtyperne af adsorptionsligevægtskurver. Adsorptionsisotermer ved tre temperaturer (T 1 > T 2 > T 3) svarer til fig. 26,9a.
Figur 26.9. Kurver for adsorptionsligevægt: isotermer (a), isobarer (b) og isostere (c) af adsorption
Ligning, der relaterer adsorptionsværdi til temperatur ved konstant ligevægtstryk A \u003d f (T) s eller konstant ligevægtskoncentration G \u003d f (T) s, kaldes henholdsvis adsorptionsisobarer eller isopycnes (fig. 26.9-b); her p 1 > p 2 > p 3. Type ligning R= f (T) A, adsorptionsisosteren (fig. 26.9-c), relaterer ligevægtstrykket til temperaturen ved en konstant adsorberet mængde; I dette tilfælde A 1 > A 2 > A 3.
Enhver adsorptionsteoris opgave er at kompilere dens matematiske beskrivelse på basis af en bestemt model af adsorptionsprocessen. Ideelt set bør ligningen beskrive afhængigheden af ligevægtsadsorptionsværdien af koncentrationen af adsorbatet i bulkfasen ved forskellige temperaturer og også forudsige ændringen i adsorptionsvarmen afhængigt af fyldningen af adsorbenten. Oftest findes adsorptionsisotermligningen i dette tilfælde. Formen af adsorptionsisotermen på faste stoffer afhænger af mange parametre: egenskaberne af adsorbenten og adsorbatet, interaktionen af adsorbentadsorbatet, interaktionen af adsorbatmolekyler med hinanden i gasfasen og i den adsorberede tilstand. I området af lave tryk (eller koncentrationer) og de tilsvarende små overfladedækninger er interaktionen mellem adsorbatmolekyler ubetydelig og afhængigheden A = f(p) T reduceret til sin enkleste form, kaldet Henriks lov:
A \u003d kp eller A \u003d k "c(26.20)
hvor k Og til"- adsorptionskoefficient (eller Henry-koefficient), fra er adsorbentkoncentrationen i bulkfasen, R er adsorbatets damptryk. Henry koefficient k er et mål for intensiteten af adsorption. Det kan påvises, at enhver teoretisk isoterm bør i grænsen (for små fyldninger) gå over i Henry-ligningen.
I regionen af medium koncentrationer, er koncentrationsafhængigheden af adsorptionen af opløste stoffer godt beskrevet af empirien Freundlich ligning:
![]() (26.21)
(26.21)
hvor x- mængden af adsorberet stof, m- adsorberende masse, βi P - konstanter, der er karakteristiske for hvert adsorptionssystem, med 0< 1/n< 1 . Ifølge Freindlich, n afhænger ikke af polstring, selvom denne erklæring ikke er helt nøjagtig. Denne empiriske ligning bruges ofte til omtrentlige beregninger af adsorption. Oftest bruges det i logaritmisk form:
hvilket gør det muligt at konstruere en lineær afhængighed ln MEN - ln c og grafisk bestemme begge konstante parametre β og n.
Overfladefænomener og adsorption. Typer af adsorptionsinteraktioner. Gasadsorptionsisotermer. Henry og Langmuirs ligning. Polymolekylær adsorption, BET-teori.
OVERFLADEFÆNOMEN OG ADSORPTION
overfladeenergi. Adsorption
Indtil nu er egenskaberne ved heterogene systemer blevet beskrevet ved hjælp af parametre og tilstandsfunktioner, der karakteriserer hver af faserne som helhed. Egenskaberne for faseområdet, der støder op til dets overflade, adskiller sig fra fasens egenskaber i volumen: faktisk danner partiklerne placeret på overfladen af hver fase en speciel overfladefase, hvis egenskaber adskiller sig væsentligt fra egenskaberne af fasens indre regioner. Partiklerne placeret på overfladen er i et andet miljø sammenlignet med partiklerne placeret i fasens volumen, dvs. interagerer både med homogene partikler og med partikler af anden art. Konsekvensen af dette er, at den gennemsnitlige energi gs for en partikel placeret på fasegrænsefladen adskiller sig fra den gennemsnitlige energi for den samme partikel i volumenet af fase gv (desuden kan energien af en partikel på overfladen være enten større eller mindre end energien af en partikel i volumenet). Derfor er den vigtigste egenskab ved overfladefasen overfladeenergi G s er forskellen mellem den gennemsnitlige energi af en partikel placeret på overfladen og en partikel placeret i fasens volumen, ganget med antallet af partikler på overfladen N:
![]() (26.1)
(26.1)
Det er indlysende, at den samlede værdi af overfladeenergien af en fase vil blive bestemt af værdien af dens overflade S. Derfor, for at karakterisere grænsefladen, der adskiller en given fase fra en anden, introduceres konceptet overfladespændingσ er forholdet mellem overfladeenergien og arealet af grænsefladen; størrelsen af overfladespændingen afhænger kun af arten af begge faser. Ligesom overfladeenergien af en fase kan overfladespændingen være enten positiv eller negativ. Overfladespændingen er positiv, hvis partiklerne på overfladen interagerer med partikler af samme fase stærkere end med partikler af en anden fase (og derfor g s > g v). Ifølge princippet om minimum fri energi vil enhver fase have tendens til spontant at reducere sin overfladeenergi; derfor, i tilfælde af positiv overfladespænding (σ > 0), har fasen en tendens til at reducere sin overflade. Hvis σ< 0, поверхностная энергия фазы будет уменьшаться при увеличении площади поверхности.
Indflydelsen af overfladelaget af en fase på dens generelle egenskaber bestemmes af andelen af partikler placeret på overfladen af det samlede antal partikler, der udgør denne fase, dvs. værdien af den specifikke overflade af fasen S/V (overflade pr. volumenhed). Den frie energi af fasen G kan repræsenteres som summen af overfladen G s og volumen G v energier proportional med fasens overfladeareal og volumen, henholdsvis:
Ved at dividere dette udtryk med fasens volumen får vi:
Det følger af ligning (IV.4), at med den samme mængde fase (dvs. konstant volumen), øges overfladeenergiens bidrag til fasens samlede energi med en stigning i det specifikke overfladeareal, eller i andre ord, grad af spredning(fragmenterings)fase. I det tilfælde, hvor spredningsgraden af fasen er lille (den specifikke overflade er ubetydelig), forsømmes normalt overfladeenergiens bidrag til fasens samlede energi. Overfladelagets bidrag til fasens og systemets egenskaber som helhed tages i betragtning ved undersøgelsen sprede systemer– heterogene systemer, hvor en af faserne er kontinuert ( dispersionsmedium), og den anden er fragmenteret ( spredt fase).
Ved grænsen af en kondenseret (dvs. fast eller flydende) fase med en gas er overfladespændingen altid positiv, da partiklerne i den kondenserede fase interagerer stærkere med hinanden end med gasmolekyler. Ifølge princippet om minimum fri energi vil den kondenserede fase have tendens til spontant at reducere sin overfladeenergi. Dette kan være resultatet af enten et fald i fasens overfladeareal (hvilket er grunden til, at en væskedråbe i vægtløshed tager form af en kugle) eller et fald i overfladespændingen, når nye partikler opstår på fasegrænsefladen - gasmolekyler eller et opløst stof. Processen med spontan ændring i koncentrationen af et stof ved grænsefladen mellem to faser kaldes adsorption. Adsorbent et stof kaldes, på hvis overflade der er en ændring i koncentrationen af et andet stof - adsorbat.
Adsorption ved opløsning-damp-grænsefladen
I flydende opløsninger er overfladespændingen σ en funktion af koncentrationen af opløst stof. På fig. 4.1 viser tre mulige afhængigheder af overfladespænding af opløsningens koncentration (de såkaldte overfladespændingsisotermer). Stoffer, hvis tilsætning til et opløsningsmiddel reducerer overfladespændingen, kaldes overfladeaktiv(overfladeaktive stoffer), stoffer, hvis tilsætning øger eller ikke ændrer overfladespændingen - overflade-inaktiv(PIAV).

Ris. 26.1 Overfladeisotermer 26.2 Adsorptionsisoterm
spænding af overfladeaktive opløsninger (1, 2) og PIAV. Overfladeaktivt middel ved opløsning-damp-grænsefladen
PIAV (3)
Et fald i overfladespænding og følgelig overfladeenergi opstår som følge af overfladeaktivt stofadsorption på væske-damp-grænsefladen, dvs. det faktum, at koncentrationen af overfladeaktivt stof i opløsningens overfladelag er større end i opløsningens dybde.
Det kvantitative mål for adsorption ved opløsning-damp-grænsefladen er overfladeoverskud G (gamma), lig med antallet af mol opløst stof i overfladelaget. Det kvantitative forhold mellem adsorptionen (overfladeoverskud) af et opløst stof og ændringen i opløsningens overfladespænding med stigende opløsningskoncentration bestemmer Gibbs adsorptionsisoterm:
Plottet af den overfladeaktive adsorptionsisoterm er vist i fig. 26.2. Det følger af ligning (26.5), at processens retning - koncentrationen af et stof i overfladelaget eller omvendt dets tilstedeværelse i hovedparten af den flydende fase - bestemmes af fortegnet af den afledte dσ / dС. Den negative værdi af denne afledte svarer til akkumuleringen af stoffet i overfladelaget (G > 0), den positive værdi svarer til en lavere koncentration af stoffet i overfladelaget sammenlignet med dets koncentration i hovedparten af opløsningen.
Værdien g = –dσ/dС kaldes også overfladeaktiviteten af det opløste stof. Overfladeaktiviteten af overfladeaktive stoffer ved en vis koncentration af C 1 bestemmes grafisk ved at tegne en tangent til overfladespændingsisotermen i punktet C = C 1 ; i dette tilfælde er overfladeaktiviteten numerisk lig med tangenten af hældningen af tangenten til koncentrationsaksen:
Det er let at se, at med stigende koncentration falder overfladeaktiviteten af overfladeaktive stoffer. Derfor bestemmes overfladeaktiviteten af et stof normalt ved en uendelig lille koncentration af opløsningen; i dette tilfælde afhænger dens værdi, betegnet g o, kun af arten af det overfladeaktive middel og opløsningsmidlet. Ved at undersøge overfladespændingen af vandige opløsninger af organiske stoffer etablerede Traube og Duclos følgende tommelfingerregel for den homologe serie af overfladeaktive stoffer:
I enhver homolog serie ved lave koncentrationer øger forlængelsen af carbonkæden med en CH2-gruppe overfladeaktiviteten med en faktor på 3-3,5.
For vandige opløsninger af fedtsyrer er overfladespændingens afhængighed af koncentrationen beskrevet af empirien. Shishkovsky ligning:
Her er b og K empiriske konstanter, og værdien af b er den samme for hele den homologiske serie, og værdien af K stiger for hvert efterfølgende medlem af serien med 3-3,5 gange.

Ris. 26.3 Begræns orienteringen af overfladeaktive molekyler i overfladelaget
Molekyler af de fleste overfladeaktive stoffer har en amfifil struktur, dvs. indeholder både en polær gruppe og en ikke-polær kulbrintegruppe. Placeringen af sådanne molekyler i overfladelaget er energimæssigt mest gunstig under den betingelse, at molekylerne er orienteret af den polære gruppe til den polære fase (polær væske), og den upolære gruppe til den ikke-polære fase (gas eller ikke-polær væske). Ved en lav koncentration af opløsningen forstyrrer termisk bevægelse orienteringen af overfladeaktive molekyler; med en stigning i koncentrationen er adsorptionslaget mættet, og et lag af "lodret" orienterede overfladeaktive molekyler dannes på grænsefladen (fig. 26.3). Dannelsen af et sådant monomolekylært lag svarer til minimumsværdien af overfladespændingen af den overfladeaktive opløsning og den maksimale værdi af adsorption G (fig. 26.1-26.2); med en yderligere stigning i koncentrationen af overfladeaktivt stof i opløsningen ændres overfladespændingen og adsorptionen ikke.
Adsorption ved fast-gas-grænsefladen
Ved adsorption af gasser på faste stoffer er beskrivelsen af vekselvirkningen mellem adsorbat- og adsorbentmolekyler et meget komplekst problem, da arten af deres vekselvirkning, som bestemmer arten af adsorption, kan være forskellig. Derfor forenkles problemet normalt ved at overveje to ekstreme tilfælde, hvor adsorption er forårsaget af fysiske eller kemiske kræfter – henholdsvis fysisk og kemisk adsorption.
fysisk adsorption opstår på grund af van der Waals interaktioner. Det er karakteriseret ved reversibilitet og et fald i adsorption med stigende temperatur, dvs. eksotermicitet, og varmeeffekten af fysisk adsorption er sædvanligvis tæt på adsorbatets flydende varme (10 – 80 kJ/mol). Sådan er for eksempel adsorptionen af inerte gasser på kul.
Kemisk adsorption(kemisorption) udføres ved kemisk interaktion mellem adsorbent- og adsorbatmolekyler. Kemisorption er normalt irreversibel; kemisk adsorption er i modsætning til fysisk adsorption lokaliseret; adsorbatmolekyler kan ikke bevæge sig over overfladen af adsorbenten. Da kemisorption er en kemisk proces, der kræver en aktiveringsenergi på omkring 40-120 kJ/mol, bidrager en temperaturstigning til dens forekomst. Et eksempel på kemisk adsorption er adsorptionen af oxygen på wolfram eller sølv ved høje temperaturer.
Det skal understreges, at fænomenerne fysisk og kemisk adsorption tydeligt skelnes i meget sjældne tilfælde. Mellemliggende muligheder udføres normalt, når hovedparten af det adsorberede stof binder relativt svagt, og kun en lille del er fast bundet. For eksempel adsorberes oxygen på metaller eller brint på nikkel ved lave temperaturer i henhold til lovene for fysisk adsorption, men efterhånden som temperaturen stiger, begynder kemisk adsorption at forekomme. Når temperaturen stiger, begynder stigningen i kemisk adsorption fra en bestemt temperatur at overlappe faldet i fysisk adsorption, så temperaturafhængigheden af adsorption i dette tilfælde har et klart defineret minimum (fig. 26.4).

Ris. 26.4 Temperaturens afhængighed af mængden af hydrogen adsorberet af nikkel
Ved en konstant temperatur afhænger mængden af adsorberet stof kun af ligevægtstrykket eller koncentrationen af adsorbatet; ligningen, der relaterer disse mængder, kaldes adsorptionsisotermen.
Teorier om adsorption
Der er ingen forenet teori, der tilstrækkeligt vil beskrive alle typer adsorption på forskellige fasegrænseflader; Lad os derfor overveje nogle af de mest almindelige adsorptionsteorier, der beskriver individuelle typer adsorption på fast-gas- eller fast-opløsning-grænsefladen.
Langmuirs teori om monomolekylær adsorption
Teorien om monomolekylær adsorption, som blev udviklet af den amerikanske kemiker I. Langmuir, er baseret på følgende bestemmelser.
1) Adsorption er lokaliseret og er forårsaget af kræfter tæt på kemiske.
2) Adsorption sker ikke på hele overfladen af adsorbenten, men på aktive centre, som er fremspring eller fordybninger på overfladen af adsorbenten, karakteriseret ved tilstedeværelsen af den såkaldte. frie valenser. Aktive centre betragtes som uafhængige (dvs. ét aktivt center påvirker ikke andres adsorptionskapacitet) og identiske.
3) Hvert aktivt center er kun i stand til at interagere med et adsorbatmolekyle; som følge heraf kan der kun dannes et lag adsorberede molekyler på overfladen.
4) Adsorptionsprocessen er reversibel og afbalanceret– det adsorberede molekyle tilbageholdes af det aktive center i nogen tid, hvorefter det desorberes; således er der efter nogen tid etableret en dynamisk ligevægt mellem processerne adsorption og desorption.

Ris. 26.5 Monomolekylær adsorptionsisoterm
I ligevægtstilstanden er adsorptionshastigheden lig med desorptionshastigheden. Desorptionshastigheden er direkte proportional med andelen af besatte aktive centre (x), og adsorptionshastigheden er direkte proportional med produktet af adsorbatkoncentrationen og fraktionen af frie aktive centre (1 – x):
![]() (26.9)
(26.9)
Herfra finder vi x:
Ved at dividere tælleren og nævneren for højre side af ligning (26.10) med k A , får vi:
 (26.11)
(26.11)
Den maksimalt mulige værdi af adsorption To opnås under den betingelse, at alle aktive centre er optaget af adsorbatmolekyler, dvs. x = 1. Derfor følger det, at x = r / r o. Sætter vi dette ind i ligning (26.11), får vi:
Ligning (26.13) er monomolekylær adsorptionsisoterm, som relaterer værdien af adsorption G til koncentrationen af adsorbat C. Her er b en vis konstant værdi for et givet adsorbent-adsorbat par (forholdet mellem desorptions- og adsorptionshastighedskonstanter), numerisk lig med adsorbatkoncentrationen, hvor halvdelen af de aktive centre er besat. Tidsplan Langmuir adsorptionsisotermer vist i fig. 26,5. Konstanten b kan bestemmes grafisk ved at tegne en tangent til adsorptionsisotermen i punktet C = 0.
Når man beskriver processen med adsorption af gasser i ligning (26.13), kan koncentrationen erstattes af en proportional værdi af gassens partialtryk:
Langmuirs teori om monomolekylær adsorption er anvendelig til at beskrive nogle processer for adsorption af gasser og opløste stoffer ved lave tryk (koncentrationer) af adsorbatet.
Polanyis teori om polymolekylær adsorption
I praksis er der ofte (især ved adsorption af dampe) såkaldte. S-formede adsorptionsisotermer (fig. 4.6), hvis form indikerer den mulige, ud fra en bestemt trykværdi, interaktionen af adsorberede molekyler med adsorbatet.

Ris. 26.6 Polymolekylær adsorptionsisoterm
For at beskrive sådanne adsorptionsisotermer foreslog M. Polyani teori om polymolekylær adsorption baseret på følgende hovedprincipper:
1. Adsorption forårsaget rent fysiske kræfter.
2. Adsorberende overflade homogen, dvs. der er ingen aktive centre på det; adsorptionskræfter danner et kontinuerligt kraftfelt nær overfladen af adsorbenten.
3. Adsorptionskræfter virker i en afstand større end størrelsen af adsorbatmolekylet. Med andre ord, på overfladen af adsorbenten er der nogle adsorptionsvolumen, som er fyldt med adsorbatmolekyler under adsorption.
4. Tiltrækningen af et adsorbatmolekyle af den adsorberende overflade afhænger ikke af tilstedeværelsen af andre molekyler i adsorptionsvolumenet, som et resultat af hvilket det er muligt polymolekylær adsorption.
5. Adsorptionskræfter ikke afhængig af temperaturen og følgelig ændres adsorptionsvolumenet ikke med en temperaturændring.
Freundlich ligning
De teoretiske koncepter udviklet af Langmuir og Polanyi idealiserer og forenkler i vid udstrækning det sande billede af adsorption. Faktisk er overfladen af adsorbenten inhomogen, der er en vekselvirkning mellem de adsorberede partikler, aktive centre er ikke helt uafhængige af hinanden osv. Alt dette komplicerer isotermligningens form. G. Freindlich viste, at ved en konstant temperatur er antallet af mol adsorberet gas eller opløst stof pr. masseenhed af adsorbenten (den såkaldte specifikke adsorption x/m) proportional med ligevægtstrykket (for gas) eller ligevægtskoncentrationen ( for stoffer adsorberet fra opløsning) af adsorbenten hævet til en vis styrke, som altid er mindre end én:
Adsorption ved grænsefladen med fast opløsning
Molekylær adsorption fra opløsninger
Adsorptionsisotermer af opløste stoffer fra en opløsning ligner i udseende adsorptionsisotermer for gasser; for fortyndede opløsninger er disse isotermer godt beskrevet af Freundlich- eller Langmuir-ligningerne, hvis ligevægtskoncentrationen af det opløste stof i opløsningen er substitueret i dem. Adsorption fra opløsninger er dog et meget mere komplekst fænomen sammenlignet med gasformigt, da adsorptionen af et opløsningsmiddel ofte sker samtidig med adsorptionen af et opløst stof.

Ris. 26.8 Orientering af overfladeaktive molekyler på den adsorberende overflade
Adsorptionens afhængighed af strukturen af adsorbatmolekyler er meget kompleks, og det er ret vanskeligt at udlede nogen regelmæssigheder. Molekyler af mange organiske stoffer består af polære (hydrofile) og ikke-polære (hydrofobe) grupper, dvs. er overfladeaktive stoffer. Når de adsorberes på en fast adsorbent, er overfladeaktive molekyler orienteret på dens overflade på en sådan måde, at den polære del af molekylet vender mod den polære fase, og den upolære del vender mod den ikke-polære fase. Så under adsorptionen af alifatiske carboxylsyrer fra vandige opløsninger på en upolær adsorbent - aktivt kul - orienteres molekylerne af carbonhydridradikaler mod adsorbenten; når det adsorberes fra benzen (ikke-polært opløsningsmiddel) på en polær adsorbent - silicagel - vil orienteringen af syremolekylerne blive omvendt (fig. 4.8).
Adsorption fra elektrolytopløsninger
Adsorption fra vandige opløsninger af elektrolytter sker som regel på en sådan måde, at ioner af samme type adsorberes fra opløsningen på den faste adsorbent. Foretrukken adsorption fra en opløsning eller en anion eller en kation bestemmes af arten af adsorbenten og ionerne. Mekanismen for adsorption af ioner fra elektrolytopløsninger kan være forskellig; allokere udveksling og specifik adsorption af ioner.
Udvekslingsadsorption er en proces med ionbytning mellem en opløsning og en fast fase, hvor den faste fase absorberer ioner af et bestemt fortegn (kationer eller anioner) fra opløsningen og i stedet frigiver et tilsvarende antal andre ioner af samme fortegn til opløsningen. Udvekslingsadsorption er altid specifik, dvs. for en given adsorbent er kun visse ioner i stand til at udveksle; udvekslingsadsorption er normalt irreversibel.
På specifik adsorption adsorption på overfladen af den faste fase af ioner af nogen art er ikke ledsaget af frigivelse i opløsningen af et tilsvarende antal andre ioner af samme fortegn; den faste fase får en elektrisk ladning. Dette fører til, at nær overfladen, under påvirkning af elektrostatiske tiltrækningskræfter, grupperes et tilsvarende antal ioner med modsatte ladninger, dvs. der dannes et elektrisk dobbeltlag. Samspillet mellem ladninger, der koncentrerer sig om overfladen, fører til et fald i systemets overfladeenergi. I tilfælde af specifik elektrolytadsorption formulerede Peskov og Fayans følgende empiriske regel ( Peskov-Fajance regel):
På overfladen af et krystallinsk fast stof adsorberes en ion specifikt fra en elektrolytopløsning, som er i stand til at fuldende sit krystalgitter eller kan danne en dårligt opløselig forbindelse med en af ionerne, der udgør krystallen.
Henrys lov kan formuleres som følger: når systemet fortyndes (trykket falder), tenderer fordelingskoefficienten til en konstant værdi, der er lig med Henrys fordelingskonstanten. Med hensyn til adsorptionsværdien A kan denne lov skrives som følger:
E  Disse ligninger er adsorptionsisotermer af et stof i lave koncentrationer. I overensstemmelse med dem kan Henrys lov formuleres som følger: mængden af adsorption ved lave gastryk (stofkoncentrationer i opløsning) er direkte proportional med trykket (koncentration).
Disse ligninger er adsorptionsisotermer af et stof i lave koncentrationer. I overensstemmelse med dem kan Henrys lov formuleres som følger: mængden af adsorption ved lave gastryk (stofkoncentrationer i opløsning) er direkte proportional med trykket (koncentration).
Afvigelser fra Henrys lov, udtrykt ved ændringer i aktivitetskoefficienterne i faserne, giver normalt ikke mulighed for at beskrive og forudsige forløbet af isotermer med stigende koncentration.
(tryk) af adsorbatet. For at opnå en teoretisk adsorptionsisoterm, der beskriver et bredere spektrum af koncentrationer, er det nødvendigt at bruge koncepter for adsorptionsmekanismen og specifikke modeller.
En stor del af afvigelser af aktivitetskoefficienten for adsorbatet i overfladelaget fra enhed kan tages i betragtning ved anvendelse af adsorptionsbegrebet som en kvasi-kemisk reaktion mellem adsorbatet og adsorptionscentrene på den adsorberende overflade. Dette er hovedideen i Langmuirs adsorptionsteori. Denne bestemmelse præciseres af følgende forudsætninger:
1) adsorption er lokaliseret (molekyler bevæger sig ikke over overfladen) på separate adsorptionscentre, som hver især interagerer med kun ét adsorbatmolekyle; som et resultat dannes et monomolekylært lag;
2) adsorptionscentre er energetisk ækvivalente - overfladen af adsorbenten er ækvipotential;
3) adsorberede molekyler interagerer ikke med hinanden.
Lyofile spredningssystemer. Klassificering og generelle karakteristika for pav. Termodynamik og micelliseringsmekanisme. Strukturen af overfladeaktive miceller i vandige og kulbrintemedier. Solubilisering.
Alle dispergerede systemer, afhængigt af mekanismen for deres dannelse, i henhold til klassificeringen af PA Rebinder, er opdelt i lyofile, som opnås ved spontan spredning af en af faserne (spontan dannelse af et heterogent frit-dispergeret system) og lyofobe som følge af dispergering og kondensation med overmætning (tvungen dannelse af et heterogent fritgående system).
Hvis overfladespændingen ved grænsefladen falder med stigende koncentration af et stof, så kaldes et sådant stof overfladeaktivt. For sådanne stoffer er overfladeaktiviteten
Tilstedeværelsen af hydrofile og oleofile dele i overfladeaktive molekyler er et karakteristisk kendetegn ved deres struktur. Ifølge evnen til at dissociere i vandige opløsninger opdeles overfladeaktive stoffer i ioniske og ikke-ioniske. Til gengæld er ioniske overfladeaktive stoffer opdelt i anioniske, kationiske og amfolytiske (amfotere).
1) Anioniske overfladeaktive stoffer dissocierer i vand for at danne en overfladeaktiv anion.
2) Kationiske overfladeaktive stoffer dissocierer i vand og danner en overfladeaktiv kation.
3) Amfolytiske overfladeaktive stoffer indeholder to funktionelle grupper, hvoraf den ene er sur og den anden basisk, såsom carboxyl- og amingrupper. Afhængigt af mediets pH udviser amfolytiske overfladeaktive stoffer anioniske eller kationiske egenskaber.
Alle overfladeaktive stoffer med hensyn til deres adfærd i vand er opdelt i virkelig opløselige og kolloide.
Virkelig opløselige overfladeaktive stoffer i opløsning er i en molekylært dispergeret tilstand op til koncentrationer svarende til deres mættede opløsninger og adskillelse af systemet i to kontinuerlige faser.
Det vigtigste kendetegn ved kolloide overfladeaktive stoffer er evnen til at danne termodynamisk stabile (lyofile) heterogene disperse systemer (associative eller micellære kolloider). De vigtigste egenskaber ved kolloide overfladeaktive stoffer, som bestemmer deres værdifulde kvaliteter og brede anvendelse, omfatter høj overfladeaktivitet; evnen til spontan micelledannelse - dannelsen af lyofile kolloide opløsninger ved en overfladeaktivt stofkoncentration over en vis specifik værdi, kaldet den kritiske micellekoncentration (KKM); evnen til at solubilisere - en kraftig stigning i opløseligheden af stoffer i opløsninger af kolloide overfladeaktive stoffer på grund af deres "introduktion" i micellerne; høj evne til at stabilisere forskellige spredningssystemer.
Ved koncentrationer over KKM opsamles overfladeaktive molekyler i miceller (associeret), og opløsningen omdannes til et micellært (associativt) kolloidt system.
En overfladeaktiv micelle forstås som en associering af amfifile molekyler, hvis lyofile grupper vender mod det tilsvarende opløsningsmiddel, og de lyofobe grupper er forbundet med hinanden og danner kernen af micellen. Antallet af molekyler, der udgør en micelle, kaldes associationstallet, og den samlede sum af molekylvægtene af molekylerne i micellen, eller produktet af massen af micellen og Avogadro-tallet, kaldes micellemassen. En vis orientering af amfifile overfladeaktive molekyler i en micelle giver en minimal grænsefladespænding ved micelle-miljøgrænsen.
P  Ved koncentrationer af overfladeaktive stoffer i en vandig opløsning, der er noget højere end KKM, dannes der ifølge Hartleys koncepter sfæriske miceller (Hartley miceller). Den indre del af Gartley-miceller består af sammenflettede kulbrinteradikaler, de polære grupper af overfladeaktive molekyler omdannes til den vandige fase. Diameteren af sådanne miceller er lig med to gange længden af overfladeaktive molekyler. Antallet af molekyler i en micelle vokser hurtigt inden for et snævert koncentrationsområde, og med en yderligere stigning i koncentrationen ændres det praktisk talt ikke, men antallet af miceller stiger. Kugleformede miceller kan indeholde fra 20 til 100 molekyler eller mere.
Ved koncentrationer af overfladeaktive stoffer i en vandig opløsning, der er noget højere end KKM, dannes der ifølge Hartleys koncepter sfæriske miceller (Hartley miceller). Den indre del af Gartley-miceller består af sammenflettede kulbrinteradikaler, de polære grupper af overfladeaktive molekyler omdannes til den vandige fase. Diameteren af sådanne miceller er lig med to gange længden af overfladeaktive molekyler. Antallet af molekyler i en micelle vokser hurtigt inden for et snævert koncentrationsområde, og med en yderligere stigning i koncentrationen ændres det praktisk talt ikke, men antallet af miceller stiger. Kugleformede miceller kan indeholde fra 20 til 100 molekyler eller mere.
Efterhånden som koncentrationen af overfladeaktivt stof stiger, passerer det micellære system gennem en række ligevægtstilstande, der adskiller sig i forbindelsesantal, størrelser og former for miceller. Når en vis koncentration er nået, begynder sfæriske miceller at interagere med hinanden, hvilket bidrager til deres deformation. Miceller har en tendens til at antage en cylindrisk, skiveformet, stangformet, lamelformet form.
Micellisering i ikke-vandige medier er som regel resultatet af virkningen af tiltrækkende kræfter mellem de polære grupper af overfladeaktive stoffer og interaktionen af carbonhydridradikaler med opløsningsmiddelmolekyler. De dannede omvendte miceller indeholder ikke-hydrerede eller hydratiserede polære grupper indeni, omgivet af et lag af kulbrinteradikaler. Associationstallet (fra 3 til 40) er meget mindre end for vandige opløsninger af overfladeaktive stoffer. Som regel vokser det med en stigning i kulbrinteradikalet op til en vis grænse.
Fænomenet med opløsning af stoffer i overfladeaktive miceller kaldes solubilisering. Måden, hvorpå solubilisatmolekyler indgår i miceller i vandige opløsninger, afhænger af stoffets beskaffenhed. Ikke-polære kulbrinter, der trænger ind i miceller, er placeret i micellers kulbrintekerner. Polære organiske stoffer (alkoholer, aminer, syrer) inkorporeres i en micelle mellem overfladeaktive molekyler, således at deres polære grupper vender mod vand, og de lipofile dele af molekylerne er orienteret parallelt med de overfladeaktive kulbrinte-radikaler. En tredje måde at inkorporere solubilisatet i miceller på er også mulig, hvilket især er karakteristisk for ikke-ioniske overfladeaktive stoffer. Molekyler af et solubilisat, såsom phenol, trænger ikke ind i miceller, men er fikseret på deres overflade, placeret mellem tilfældigt bøjede polyoxyethylenkæder.
Solubilisering er en spontan og reversibel proces; en given overfladeaktivt stofkoncentration og temperatur svarer til en veldefineret mætning af opløsningen med solubilisat. Som et resultat af solubilisering opnås stabile disperse systemer svarende til spontant dannede ultramikroheterogene emulsioner.
Bestem overfladen og den samlede (indre) energi af 4 g vandtåge, som har partikler med en spredning på 5 10 7 m -1 , t= 20ºCσ = 72 mJ/m 2 ; dσ/ dT= -0,16 mJ/(m 2 ·TIL); ρ = 1000 kg/m 3 .

Eksamensbillet nummer 10
Teori om polymolekylær BET-adsorption: startpositioner, afledning af isotermligningen og dens analyse. Lineær form af BET-ligningen. Bestemmelse af den specifikke overflade af adsorbenter, katalysatorer og andre porøse legemer.
Langmuir-ligningen kan kun bruges under den betingelse, at adsorptionen af et stof er ledsaget af dannelsen af et monomolekylært lag.
I de fleste tilfælde kompenserer det monomolekylære adsorptionslag ikke fuldt ud for den overskydende overfladeenergi, og påvirkningen af overfladekræfter kan strække sig til det andet, tredje og efterfølgende adsorptionslag, hvilket resulterer i polymolekylær adsorption.
FRA  Den moderne form for den polymolekylære adsorptionsligning - den grundlæggende ligning i den generaliserede Langmuir-teori - blev foreslået af Brunauer, Emmett og Teller.
Den moderne form for den polymolekylære adsorptionsligning - den grundlæggende ligning i den generaliserede Langmuir-teori - blev foreslået af Brunauer, Emmett og Teller.
I denne teori er en yderligere antagelse til dem, der blev brugt som grundlag for udledningen af Langmuir-isotermligningen, ideen om dannelsen på overfladen af adsorbenten af "på hinanden følgende komplekser" af adsorptionscentre med en, to, tre osv. molekyler af adsorbatet. Derefter kan adsorptionsprocessen repræsenteres som successive kvasi-kemiske reaktioner:
Ligevægtskonstanterne for disse reaktioner er henholdsvis lig med
Angiv:
Det samlede antal aktive steder på adsorbenten, eller kapaciteten af monolaget, vil være lig med

Efter en række beregninger ved hjælp af serieteorien får vi endelig:

D  Denne relation er den grundlæggende ligning i den generaliserede Langmuir-teori og kaldes BET polymolekylær adsorptionsligning.
Denne relation er den grundlæggende ligning i den generaliserede Langmuir-teori og kaldes BET polymolekylær adsorptionsligning.
Ved behandling af eksperimentelle resultater bruges BET-ligningen normalt i en lineær form:
Det giver dig mulighed for grafisk at bestemme både konstante parametre A ∞ og С:
Den eksperimentelle bestemmelse af A ∞ gør det muligt at beregne det specifikke overfladeareal af adsorbenten (overfladeareal pr. masseenhed af adsorbenten): ![]() .
.
Adsorption- koncentration af et stof fra mængden af faser på grænsefladen mellem dem. Adsorption kan ses som absorption substans (adsorbat) overflade af adsorbenten.
Adsorbent Stoffet på hvis overflade adsorption finder sted.
Adsorbtiv - en gas eller et opløst stof, der er i stand til at blive adsorberet på overfladen af en adsorbent.
Adsorbat - adsorberet stof på overfladen af adsorbenten. Ofte identificeres begreberne "adsorbent" "adsorbat".
Skelne fysisk adsorption, sker uden kemisk ændring af adsorbatet og kemisk adsorption(kemisorption), ledsaget af kemisk interaktion mellem adsorbenten og adsorbenten.
Adsorption sker ved fasegrænserne: fast - væske, fast - gas, væske - gas, væske - væske.
Når et stof adsorberes i form af molekyler, kaldes det molekylær adsorption i form af ioner - ionisk adsorption.
Adsorption er reversibel, den omvendte proces kaldes desorption.
Adsorptions- og desorptionshastighederne er lig med hinanden ved adsorptionsligevægt, hvilket svarer til ligevægtskoncentration adsorbat i opløsning el ligevægtstryk i gasfasen.
Adsorptionsværdi(A) er karakteriseret ved ligevægtsmængden af absorberet stof (X) pr. masseenhed fast adsorbent (m): [mol/kg eller kg/kg]
Adsorptionsisoterm- grafisk fremstilling af adsorptionsværdiens afhængighed af ligevægtskoncentrationen eller ligevægtstrykket ved en given konstant temperatur.
Skelne adsorption monomolekylær, hvor adsorbatet dækker overfladen af adsorbenten med et lag et molekyle tykt og polymolekylær, hvor adsorbatmolekylerne kan placeres på overfladen af adsorbenten i flere lag.
Monomolekylær adsorptionsisoterm har formen vist i fig. 12 ( Langmuir isoterm)
A Site I - svar lille ligevægtskoncentrationer (tryk), når en lille del af adsorbentoverfladen er optaget af adsorbatmolekyler, og afhængigheden A - c (p) er lineær;
Afsnit II - medium koncentrationer (tryk), ved hvilke en betydelig del af adsorbentoverfladen er optaget af adsorbatmolekyler;
c (p) Afsnit III - observeret kl høj ligevægtskoncentrationer (tryk), når hele adsorbentoverfladen er optaget af adsorbatmolekyler og nås grænseværdi for adsorption (A).
Isoterm af monomolekylær adsorptionsbrønd er beskrevet af Langmuir-ligningen:

hvor c, A individuelle konstanter for hvert enkelt stof under adsorption på en specifik adsorbent;
s, r- ligevægtskoncentration eller ligevægtstryk.
Ved lave ligevægtskoncentrationer kan vi negligere værdien fra eller R i nævneren. Derefter transformeres Langmuir-ligningen til ligningen for en ret linje, der går gennem oprindelsen:
A = A i c eller A \u003d A i s
På høje ligevægtskoncentrationer kan negligeres i nævneren i. Derefter transformeres Langmuir-ligningen til ligningen for en ret linje uafhængig af fra eller R: A = A
Til praktiske beregninger det er nødvendigt at kende konstanterne for Langmuir-ligningen A og i. At transformere ligningen til en lineær form af en ret linje, der ikke går gennem koordinaternes oprindelse: , giver dig mulighed for at bygge en graf over afhængigheden 1/A - 1/c (fig. 13).
1/A Segmentet OB er lig med 1/A. Koefficient i kan findes ud fra det faktum, at i er lig med den koncentration, ved hvilken mængden af adsorption er halvdelen af grænsen.

På grafen bestemmer interpolation segmentet OD svarende til 2/A og lig med 1/in. Så er s = 1/OD.
Langmuir-ligningen blev afledt af teorien om monomolekylær adsorption, som har følgende grundlæggende bestemmelser:
adsorption af molekyler forekommer kun på adsorptionscentre (toppe af uregelmæssigheder og smalle porer);
hvert adsorptionscenter kan kun indeholde ét adsorbatmolekyle;
adsorptionsprocessen er reversibel; Adsorptionsligevægt er dynamisk. Adsorberede molekyler tilbageholdes kun af adsorptionscentre i en vis tid, hvorefter disse molekyler desorberes og det samme antal nye molekyler adsorberes.
Ud over Langmuir-ligningen bruges den i praksis ofte Freundlich ligning:
A \u003d KS 1 / n eller A \u003d KR 1 / n, hvor K og 1 / n er empiriske konstanter.
Ligningen er mere egnet til at beskrive adsorption på den porøs eller pulveriseret adsorbenter i området gennemsnitlige koncentrationer (tryk).
Freundlich-adsorptionsisotermen har ikke en vandret lige linje, og adsorptionen stiger med stigende koncentration (tryk) (fig. 14).

Ris. fjorten
Til at finde konstanterne i Freundlich-ligningen den konverteres ved hjælp af logaritmen til ligningen for en ret linje, der ikke går gennem oprindelsen: lg A \u003d log K + 1 / n lg C.
I overensstemmelse hermed har grafen over lg A's afhængighed af lg C eller (P), bygget i henhold til eksperimentelle data, den form, der er vist i fig. 15. Ved ekstrapolering til ordinataksen opnås et segment OB lig med lg K. Tangensen af hældningsvinklen for den rette linie BN til abscisseaksen er 1/n ( tg =)
Polymolekylær adsorption- observeret under adsorption på porøse eller pulveriserede adsorbenter (silicagel, aktivt kul, pulvere og tabletter af medicinske stoffer). I dette tilfælde fortsætter adsorptionen, indtil der dannes et tæt monomolekylært lag, som vist i fig. 16.

Ris. 16.

En sådan adsorption svarer til en anden type isoterm (fig. 17), den såkaldte " S - isoterm".
kapillær kondensation- fænomenet damp fortætning i porerne eller kapillærerne i en fast adsorbent, det observeres, når let flydende gasser eller dampe (f.eks. vand, benzen osv.) absorberes som et resultat af polymolekylær adsorption. Hvori polymolekylært lag repræsenterer tynd film af væske dækker den indre overflade af poren. Lag af en sådan væske fusionerer med hinanden på smalle steder danner konkave menisker, under hvilke dannes damptryk. Derved porer trække gasmolekyler (damp) ind og fyldt med væske dannet under kondens.
Når det flyder adsorption kompliceret af kapillær kondensation, den isoterm, der svarer til fyldningen af porer (1), falder ikke sammen med den isoterm (2), der svarer til deres tømning (fig. 18). På isotermen, kondens hysterese loop. Adsorptions- og desorptionsprocesserne falder ikke sammen.
