Henry's wet van adsorptie. Langmuir-adsorptie-isothermvergelijking. Lyofiele dispersiesystemen. Classificatie en algemene kenmerken van pauwinnen. Thermodynamica en mechanisme van micellisatie. Structuur van micellen van oppervlakteactieve stoffen in waterige en koolwaterstofmedia. Oplossen
In het geval van interactie tussen twee atomen:
U – interactie-energie;
U = U VOORAFGAAND. + U RETOUR
 - Lennard-Jones-vergelijking
, c, b, m = constant
- Lennard-Jones-vergelijking
, c, b, m = constant
In gevallen van interactie van atomen met een vast oppervlak is het noodzakelijk om alle interacties samen te vatten.
x – afstand tot oppervlak
r – actieradius van aantrekkingskrachten
dV – volume
n – aantal oppervlaktemoleculen
U ADVERTENTIES. – energie van adsorptie-interactie



Bij adsorptie neemt de aantrekkingskracht toe. En in het geval van niet-polaire-niet-polaire interactie is de adsorptie voornamelijk gelokaliseerd in uitsparingen.
Elektrostatische interactie.
Polair adsorbens – niet-polair adsorbaat
Niet-polair adsorbens – polair adsorbaat
Polair adsorbens – polair adsorbaat.
M  Het adsorbaatmolecuul wordt weergegeven als een dipool, en het adsorbens wordt weergegeven als een geleider waarin het adsorbaatmolecuul een dipoolspiegel induceert, symmetrisch ten opzichte van de gegeven spiegel.
Het adsorbaatmolecuul wordt weergegeven als een dipool, en het adsorbens wordt weergegeven als een geleider waarin het adsorbaatmolecuul een dipoolspiegel induceert, symmetrisch ten opzichte van de gegeven spiegel.
X – afstand tot het midden
Bij interactie ontstaat potentieel:
 ,
,
 - dipoolmoment.
- dipoolmoment.
Het potentieel heeft de neiging de maximale waarde aan te nemen, d.w.z. dipolen hebben de neiging zich loodrecht op het oppervlak te oriënteren.
Omdat een temperatuurstijging de groei van de Brownse beweging bevordert, leidt dit tot remming van het adsorptieproces.
Bij elektrostatische interactie bevindt het adsorbaat zich hoofdzakelijk op de uitsteeksels.
Fundamentele adsorptievergelijking.
Bij adsorptie vindt er een herverdeling van de component plaats, waardoor de chemische potentiaal verandert. Het adsorptieproces kan worden beschouwd als de overgang van oppervlakte-energie naar chemische energie.
Laagvolume = 0, dan de algemene vergelijking van de I- en II-wetten van de thermodynamica:
T = constant; (1) = (2) => 

Voor een tweecomponentensysteem:
,
 ,
,
 =>
=>
=>
 - Gibbs-adsorptievergelijking
.
- Gibbs-adsorptievergelijking
.
In het geval van tv-adsorptie. lichaam - gas: , 
 ,
,

 - isotherm
- isotherm
 - isobaar
- isobaar
 - isopycnaal
- isopycnaal
 - isosteer
- isosteer
Isotherm, isopycne, isostere zijn aan elkaar gerelateerd.
Omdat adsorptie functie 



Henry-isotherm Langmuir-isotherm



Thermodynamica. Adsorptie.
Voor gecondenseerde materie:
 ,
,
 ,
,
 - integrale verandering in Gibbs-energie
.
- integrale verandering in Gibbs-energie
.

P – druk over een gebogen oppervlak, Р S – druk over een vlak oppervlak
 - adsorptiepotentieel
- adsorptiepotentieel
Differentiële verandering in entrapy 
 , Г = const
, Г = const
- differentiële entropieverandering
- differentiële enthalpie van adsorptie
 - isosterische adsorptiewarmte
- isosterische adsorptiewarmte
 - hitte van condensatie
- hitte van condensatie
 - netto adsorptiewarmte
- netto adsorptiewarmte

 ,
,




Qa – integrale adsorptiewarmte, 
Qra – integrale netto adsorptiewarmte,

Henry's vergelijking
De studie van adsorptie wordt gecompliceerd door de heterogeniteit van het oppervlak, dus de eenvoudigste wetten worden verkregen voor homogene oppervlakken.
Laten we eens kijken naar de interactie van gassen met een vast oppervlak, wanneer een gas overgaat van een evenwichtstoestand in het volume naar een evenwichtstoestand op het oppervlak. Dit geval is analoog aan het evenwicht van gassen in een zwaartekrachtveld.
 ,
,
 ,
=>
,
=> -Henry's vergelijking
-Henry's vergelijking
 - verdelingscoëfficiënt
- verdelingscoëfficiënt
Tijdens het adsorptieproces vindt er een verandering in chemische potentiëlen plaats.
Voor de bulkfase: 
Voor gas op het oppervlak: 
In een staat van evenwicht  , d.w.z.
, d.w.z.

In de vergelijking van Henry is de constante niet afhankelijk van de concentratie
De vergelijking van Henry is geldig in het gebied van lage drukken en concentraties. Naarmate de concentratie toeneemt, zijn er 2 soorten afwijkingen van de wet van Henry mogelijk:
1 – positieve afwijkingen, D neemt af, A neemt af
2 – negatieve afwijkingen, D – toename, A – toename.
Het type afwijking wordt bepaald door de overheersing van een of ander type adsorbens-adsorbaatinteractie.
Bij een sterke lijminteractie nemen de activiteitscoëfficiënten toe - een positieve afwijking. Bij samenhangende interacties worden negatieve afwijkingen waargenomen.
Monomoleculaire adsorptie.
Langmuir-isotherm.
De eenvoudigste patronen werden verkregen in de theorie van Henry. Langmuir stelde een theorie voor volgens welke adsorptie wordt beschouwd als een quasi-chemische reactie. Waarin:
Het oppervlak is energetisch homogeen.
Adsorptie is gelokaliseerd, elk adsorptiecentrum interageert met één adsorbaatmolecuul.
Adsorbaatmoleculen hebben geen interactie met elkaar.
Monolaag-adsorptie.

 - oppervlak,
- oppervlak,  - adsorberen,
- adsorberen,  - adsorptiecomplex.
- adsorptiecomplex.
 , dan de concentratie van adsorptieplaatsen:
, dan de concentratie van adsorptieplaatsen:  ,
, - beperking van adsorptie.
- beperking van adsorptie.
 , dan is de reactieconstante:
, dan is de reactieconstante: 

 - Langmuir-vergelijking.
- Langmuir-vergelijking.
Afhankelijkheid van adsorptie van concentratie
1 )
)
 ,
,
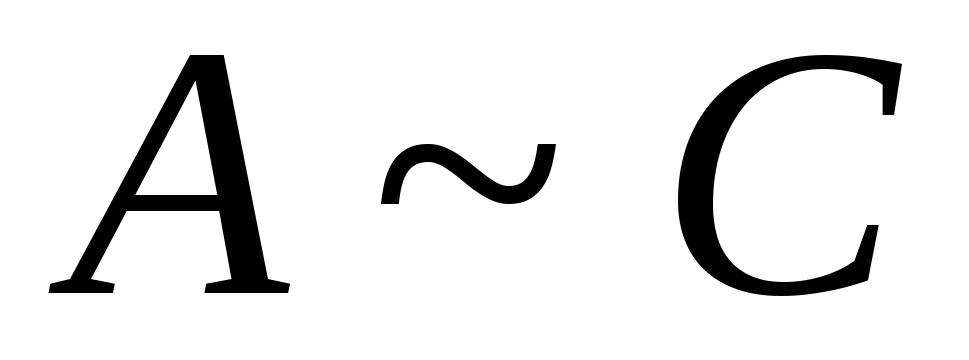
2) gebied met hoge concentraties
 - beperking van adsorptie, vorming van een monomoleculaire laag
- beperking van adsorptie, vorming van een monomoleculaire laag
Voor Gibbs-energie: .
g is de entropiefactor.
In het geval van de Henry-isotherm karakteriseert de Gibbs-energie de overgang van het adsorbaat van de standaardtoestand in de bulk naar de standaardtoestand op het oppervlak. In het geval van de Langmuir-isotherm  karakteriseert de mate van affiniteit tussen het adsorbens en het adsorbaat.
karakteriseert de mate van affiniteit tussen het adsorbens en het adsorbaat.
 gevonden van de isobaar van Van't Hoff.
gevonden van de isobaar van Van't Hoff.

 , Dan
, Dan  , vanaf hier
, vanaf hier  .
.

 - mate van oppervlaktevulling.
- mate van oppervlaktevulling.




 - aantal vrije zitplaatsen,
- aantal vrije zitplaatsen,  - aantal bezette plaatsen.
- aantal bezette plaatsen.
 ,
,

Die. in het gebied met hoge concentraties is het aantal vrije plaatsen omgekeerd evenredig met de hoeveelheid adsorbaat.
Adsorptie van een mengsel van gassen op een homogeen oppervlak.
In dit geval wordt het adsorptieproces beschouwd als twee parallelle reacties.
 (1)
(1)

 (2)
(2)


Adsorptie van een mengsel van gassen op een niet-uniform oppervlak.
Bij een niet-uniform oppervlak kan men zich niet beperken tot gemiddelde vullingen.
Als gevolg van concurrentie is lokalisatie van verschillende adsorbaten mogelijk in gebieden van verschillende typen.
In dit geval de relatie  .
.
 ,
,
 - verzadigde dampdruk van het adsorbaat.
- verzadigde dampdruk van het adsorbaat.
 ,
,
 - adsorptiewarmte.
- adsorptiewarmte.
“+” - symbaat-afhankelijkheid, “-” - anti-afhankelijkheid, “N” - geen correlatie.
“+” - adsorptie verloopt via hetzelfde mechanisme. In de energetisch meest gunstige gebieden wordt overwegend gas met een hoge affiniteit voor het oppervlak geadsorbeerd.
“-” - adsorptie vindt plaats via verschillende mechanismen en tot een bepaald moment is er geen concurrentie om het oppervlak.
Monomoleculaire adsorptie wordt voornamelijk gerealiseerd tijdens de fysische adsorptie van gassen bij lage waarden P, evenals op het grensvlak vloeistof/gas.
Polymoleculaire adsorptie.
BET-theorie(Brunauer, Emmett, Teller).
In het geval dat de vorming van een monolaag niet voldoende is om de oppervlakte-energie te compenseren, is adsorptie polymoleculair en kan worden beschouwd als een resultaat van geforceerde condensatie onder invloed van oppervlaktekrachten.
Belangrijkste punten:
Wanneer een adsorbaatmolecuul een bezette plaats raakt, wordt een meervoudige set gevormd.
Naarmate we dichterbij komen P Naar P S het aantal vrije adsorptieplaatsen neemt af. Aanvankelijk neemt het aantal plaatsen bezet door enkelspel, dubbelspel, enz. toe en vervolgens af. in sets.
Bij P =P S adsorptie verandert in condensatie.
Er zijn geen horizontale interacties.
Voor de eerste laag wordt voldaan aan de Langmuir-isotherm.
Het oppervlak wordt beschouwd als een reeks adsorptieplaatsen. De voorwaarde van dynamisch evenwicht is geldig: de condensatiesnelheid op vrije plaatsen is gelijk aan de verdampingssnelheid op bezette plaatsen.
a is de condensatiecoëfficiënt (de fractie van moleculen gecondenseerd op het oppervlak);
 ,
,
Zm – maximaal aantal vrije zitplaatsen.
 - frequentie van atomaire trillingen in de richting loodrecht op het oppervlak.
- frequentie van atomaire trillingen in de richting loodrecht op het oppervlak.
Voor de eerste laag, dynamische evenwichtsomstandigheden:

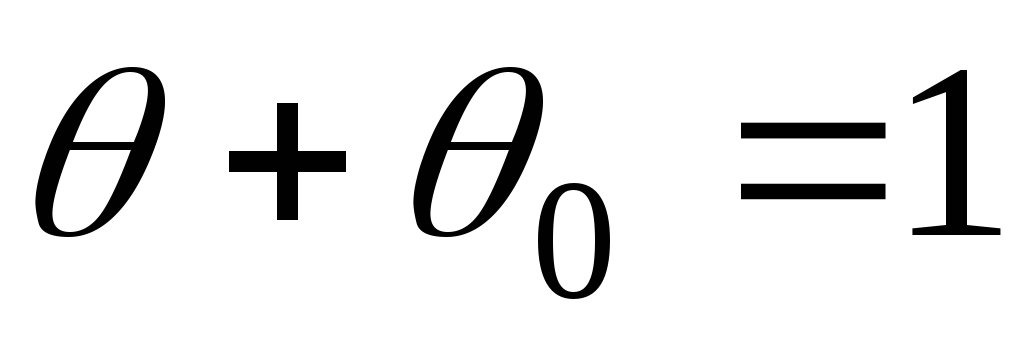

 , Dan
, Dan
 - Langmuir-vergelijking.
- Langmuir-vergelijking.
Voor de tweede laag geldt: 
Voor de i-de laag: 
Voor de eenvoud wordt aangenomen dat a en v hetzelfde zijn voor alle lagen behalve de eerste. Voor alle lagen behalve de eerste is de adsorptiewarmte constant. Voor de laatste laag is de adsorptiewarmte gelijk aan de condensatiewarmte. Als resultaat werd de vergelijking verkregen
 (*)
(*)
C– constant,
In het geval van de BET-theorie is dit de constante MET karakteriseert de Gibbs-energie van pure adsorptie. De vergelijking bevat slechts één constante, en deze vergelijking is ook erg belangrijk voor het bepalen van het specifieke oppervlak van het adsorbens.
Omdat bij adsorptie warmte vrijkomt, worden bij lage temperaturen specifieke oppervlakten bepaald.
 ????????????
????????????


Het belangrijkste nadeel van de theorie– het verwaarlozen van horizontale interacties ten gunste van verticale.
De vergelijking geldt binnen het bereik  van 0,05 tot 0,3.
van 0,05 tot 0,3.
Waar  <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 > 0,3 – de adsorbaat-adsorbaat-interactie wordt beïnvloed.
> 0,3 – de adsorbaat-adsorbaat-interactie wordt beïnvloed.
Rekening houden met adsorbaat-adsorbaatinteracties.
Interacties vinden plaats wanneer vertakte moleculen of moleculen worden geadsorbeerd op een niet-polair oppervlak. In staat om vennoten te vormen. In dit geval verandert de vorm van de adsorptie-isothermen.
A  het adsorbens is niet polair.
het adsorbens is niet polair.
Grafiek 1 komt overeen met zwakke adsorbaat-adsorbaat-interacties en sterke adsorbaat-adsorbens-interacties.
Grafiek 2 komt overeen met sterke adsorbaat-adsorbaat- en sterke adsorbaat-adsorbens-interacties.
Grafiek 3 komt overeen met een sterke adsorbaat-adsorbaat-interactie en een zwakke adsorbaat-adsorbens-interactie.
 ,
,

In het geval van interactie tussen adsorbaatmoleculen is het noodzakelijk rekening te houden met veranderingen in activiteitscoëfficiënten. En deze vergelijking wordt geschreven als:
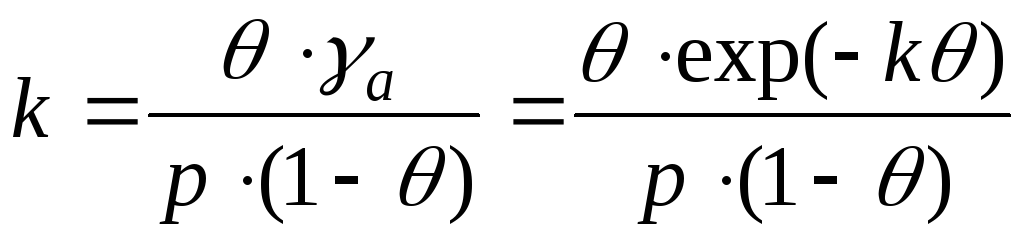 - Frunkin, Fowler, Guggenheim-vergelijking.
- Frunkin, Fowler, Guggenheim-vergelijking.
k– aantrekkingsconstante.
Polyany's potentiële theorie.
Deze theorie leidt geen enkel type adsorptie-isotherm af, maar maakt het mogelijk om isothermen bij een andere temperatuur te berekenen.
Adsorptie- dit is het resultaat van de aantrekking van het adsorbaat naar het oppervlak van het adsorbens als gevolg van de werking van het adsorptiepotentieel, dat niet afhankelijk is van de aanwezigheid van andere moleculen en afhankelijk is van de afstand tussen het oppervlak en het adsorbaatmolecuul.
 ,
,
 - adsorptiepotentieel.
- adsorptiepotentieel.
Omdat het oppervlak niet-uniform is, wordt de afstand vervangen door het adsorptievolume  .Adsorptievolume is het volume dat is ingesloten tussen het oppervlak en het punt dat overeenkomt met een gegeven waarde
.Adsorptievolume is het volume dat is ingesloten tussen het oppervlak en het punt dat overeenkomt met een gegeven waarde  .
.
Adsorptiepotentieel is het werk van het overbrengen van 1 mol adsorbaat buiten een bepaald adsorptievolume naar een bepaald punt van het adsorptievolume (of het overbrengen van 1 mol verzadigde damp van een adsorbaat dat in evenwicht is met een vloeibaar adsorbaat bij afwezigheid van een adsorbens in een dampfase in evenwicht met het adsorbens).

Karakteristieke curve
 - adsorptiepotentieel,
- adsorptiepotentieel, 
Voor een bepaald adsorbens en verschillende adsorbaten geldt het volgende: 
Voor verschillende soorten adsorbaten  ,
,
Waar  mogelijkheden voor adsorptie-isothermen bij relatieve drukken
mogelijkheden voor adsorptie-isothermen bij relatieve drukken  voor adsorbaat 1 en voor adsorbaat 2. Deze verhouding is een constante waarde.
voor adsorbaat 1 en voor adsorbaat 2. Deze verhouding is een constante waarde.
 - affiniteitscoëfficiënt
- affiniteitscoëfficiënt
Theorie van capillaire condensatie.
Het verloop van het adsorptieproces hangt grotendeels af van de structuur van het poreuze lichaam.
|
Microporeus | |
|
Transitioneel poreus | |
|
Macroporeus |
In het geval van microporeuze sorptiemiddelen overlappen de velden van adsorptiekrachten elkaar. In het geval van macroporeuze sorptiemiddelen fungeren poriën als transportkanalen. Condensatieprocessen zijn het belangrijkst in transitioneel poreuze lichamen. Capillaire condensatie begint bij bepaalde waarden P En  , wanneer een deel van de oppervlakte-energie al is gecompenseerd. Een noodzakelijke voorwaarde is dat de ondergrond zelfbevochtigend is. Het proces wordt beschreven Thompson-Kelvin-vergelijking.
, wanneer een deel van de oppervlakte-energie al is gecompenseerd. Een noodzakelijke voorwaarde is dat de ondergrond zelfbevochtigend is. Het proces wordt beschreven Thompson-Kelvin-vergelijking.

 - bij bevochtiging ligt het krommingsmiddelpunt in de gasfase.
- bij bevochtiging ligt het krommingsmiddelpunt in de gasfase.
Bij capillaire condensatie heeft de adsorptie-isotherm een hysteretische vorm. De onderste tak komt overeen met het adsorptieproces en de bovenste tak komt overeen met het desorptieproces.
Alle soorten poriën kunnen worden teruggebracht tot drie typen:
|
Conisch |
Cilindrisch met één gesloten uiteinde |
Cilindrisch met twee open uiteinden |
|
Procesvulling wordt uitgevoerd vanaf de bodem van de porie. De adsorptie-isotherm en desorptie-isotherm vallen in dit geval samen, aangezien het adsorptieproces begint vanuit een bol en het desorptieproces ook begint met het verdwijnen van enkele bollen.
↓ |
Er is geen hysteresis. Voorwaartse en achterwaartse slag worden beschreven door de vergelijking:
|
Er is nergens een bodem, de vulling van de porie zal langs de wanden van de cilinder gaan.
cilinder: De isotherm zal een hysteretisch uiterlijk hebben.
↓ |


 - bol,
- bol, ,
,



IN  Onder bevochtigingsomstandigheden treedt condensatie op bij lagere drukken, wat energetisch gunstig is. Vanuit de desorptietak worden curven van de poriegrootteverdeling verkregen.
Onder bevochtigingsomstandigheden treedt condensatie op bij lagere drukken, wat energetisch gunstig is. Vanuit de desorptietak worden curven van de poriegrootteverdeling verkregen.
Het maximum van de differentiële curve wordt naar links verschoven ten opzichte van het buigpunt van de integrale curve. Het totale volume kleine poriën is klein, maar heeft grote oppervlakken. Met toenemende poriegrootte neemt hun volume toe  , en de omgeving is zo
, en de omgeving is zo  Hierdoor wordt een verschuiving in het maximum van de differentiële curve waargenomen.
Hierdoor wordt een verschuiving in het maximum van de differentiële curve waargenomen.
Adsorptie aan het vast-vloeistof grensvlak.
In het geval van adsorptie aan het grensvlak tussen vast en gas hebben we één component verwaarloosd. In het geval van adsorptie aan het vast-vloeistofgrensvlak verdringt het adsorbaat oplosmiddelmoleculen van het oppervlak van het adsorbens.
 ,
,
De vergelijking klopt:

 ,
,
N 1, N 2 – molfracties van oplosmiddel en component, N 1 + N 2 = 1, dan
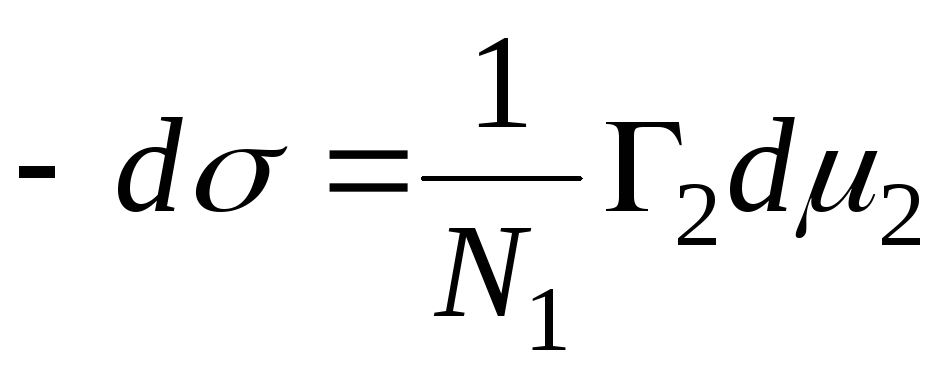 ,
=>
,
=>
 , dan is de adsorptievergelijking voor het vaste-vloeistofgrensvlak.
, dan is de adsorptievergelijking voor het vaste-vloeistofgrensvlak.
Adsorptie (G) > 0 at  <
0
<
0
Als de waarden  want de component en het oplosmiddel zijn heel verschillend, in dit geval de afhankelijkheid G van N heeft een extremum bij de waarde N
~ 0,5.
want de component en het oplosmiddel zijn heel verschillend, in dit geval de afhankelijkheid G van N heeft een extremum bij de waarde N
~ 0,5.
E  als
als  hebben nauwe waarden, in dit geval kan het teken van adsorptie veranderen. Verslaving G van N kruist de x-as
hebben nauwe waarden, in dit geval kan het teken van adsorptie veranderen. Verslaving G van N kruist de x-as
Functie snijpunt G(N) met de x-as wordt aangeroepen adsorptie azeotroop. Dit betekent dat de twee componenten op een bepaald adsorbens niet kunnen worden gescheiden.
Vergelijking van adsorptie-isotherm met uitwisselingsconstante.
Tijdens adsorptie aan het grensvlak tussen vaste stof en vloeistof vindt er een constante herverdeling van componenten plaats tussen het oppervlak van het adsorbens en het volume van de oplossing.
 - componenten (- - verwijzen naar het oppervlak)
- componenten (- - verwijzen naar het oppervlak)

 ,
,
 ,
, .
.
 ,
,


Adsorptie aan het vloeistof-gasgrensvlak
R  Laten we eens kijken naar de verandering in het concentratieprofiel wanneer het grensvlak tussen vloeistof en gas wordt overschreden. Laat component 2 vluchtig zijn.
Laten we eens kijken naar de verandering in het concentratieprofiel wanneer het grensvlak tussen vloeistof en gas wordt overschreden. Laat component 2 vluchtig zijn.
Cs – concentratie in de oppervlaktelaag.
Gebaseerd op de definitie van overmatige adsorptie

Als de component niet vluchtig is, wordt de adsorptiewaarde als volgt geschreven:
P  Ri
Ri 

In vgl.  de aard van een stof wordt beschreven door zijn derivaat
de aard van een stof wordt beschreven door zijn derivaat  .
.
De oppervlaktespanningsisotherm kan de vorm 1 of 2 hebben:
1 – oppervlakteactieve stoffen
2 – oppervlakteactieve stoffen
Oppervlakteactiviteit g is het vermogen van stoffen om de oppervlaktespanning in een systeem te verminderen.

 - dikte van de oppervlaktelaag
- dikte van de oppervlaktelaag
C S– concentratie van de component in de oppervlaktelaag
MET– volumeconcentratie
Voor een homologe reeks geldt een regel:
 - Traubo Duclos-heerschappij
- Traubo Duclos-heerschappij
Voor een homologe reeks ziet de adsorptie-isotherm er als volgt uit:
In plaats van A schrijven we G, omdat de adsorptie in de oppervlaktelaag excessief is.
Oppervlaktespanning isotherm:

 - oppervlaktespanning van een zuiver oplosmiddel.
- oppervlaktespanning van een zuiver oplosmiddel.
 - fundamentele adsorptievergelijking;
- fundamentele adsorptievergelijking;
 - Langmuir-vergelijking.
- Langmuir-vergelijking.
Laten we ze samen oplossen:

- Shishkovsky-vergelijking.
B– constante voor de homologe reeks.
A- bij het overstappen van de ene homoloog naar de andere neemt het 3-3,5 keer toe 
![]()
1 – gebied met lage concentraties
![]()
2 – gemiddelde concentratie
3 – monomoleculaire laag
Oppervlakteactieve stoffen zijn difiele moleculen, d.w.z. omvatten een polaire groep en een niet-polair koolwaterstofradicaal.
o is het polaire deel van het molecuul.
| - niet-polair deel van het molecuul.
In een polair oplosmiddel zijn oppervlakteactieve moleculen zo georiënteerd dat het polaire deel van het molecuul naar het oplosmiddel is gericht en het niet-polaire deel in de gasfase wordt geduwd.
In de vergelijking van Shishkovsky  , het is constant voor de homologische reeks.
, het is constant voor de homologische reeks.
Het oppervlakteactieve effect begint te verschijnen N>5. Bij concentraties hoger dan de concentratie van de monomoleculaire laag treedt micellisatie op in oplossingen van oppervlakteactieve stoffen.
Micel– wordt een aggregaat van amfifiele oppervlakteactieve moleculen genoemd, waarvan de koolwaterstofradicalen een kern vormen en de polaire groepen in de waterfase worden omgezet.
Micellaire massa – micellaire massa.
H  aantal moleculen – aggregatienummer.
aantal moleculen – aggregatienummer.
Bolvormige micellen
Bij micellisatie ontstaat er een evenwicht in de oplossing
CMC – kritische concentratie van micelvorming.
Omdat we de micel als een aparte fase beschouwen:
Voor een homologische reeks is er een empirische vergelijking:
A– energie van ontbinding van de functionele groep.
B – toename van het adsorptiepotentieel, adsorptie-arbeid per methyleeneenheid.
– toename van het adsorptiepotentieel, adsorptie-arbeid per methyleeneenheid.
De aanwezigheid van een koolwaterstofkern in micellen creëert de mogelijkheid dat verbindingen die onoplosbaar zijn in water worden opgelost in waterige oplossingen van oppervlakteactieve stoffen; dit fenomeen wordt solubilisatie genoemd (wat oplost is het solubilisaat, de oppervlakteactieve stof is het solubilisator).
De modder kan volledig niet-polair zijn, kan zowel polaire als niet-polaire delen bevatten en zal georiënteerd zijn als een oppervlakteactief molecuul.
In ieder geval is er tijdens het solubiliseren een toename van de micellaire massa en het aggregatiegetal, niet alleen als gevolg van de opname van solubilisaat, maar ook als gevolg van een toename van het aantal oppervlakteactieve moleculen dat nodig is om een evenwichtstoestand te handhaven.
Oplossen is effectiever naarmate het molecuulgewicht van het oplosbaarheidsmiddel lager is.

 ~ 72 mN\m.
~ 72 mN\m.
 ~ 33 mN\m.
~ 33 mN\m.
De effectiviteit van oppervlakteactieve stoffen is afhankelijk van de CMC-waarde.
2D-oppervlaktelaagdruk

→ -oppervlaktespanningskrachten.
- tweedimensionale druk.
De oppervlaktelaag is een kracht gelijk aan het verschil in oppervlaktespanning van een oppervlakteactieve oplossing en een puur oplosmiddel, gericht op een schoon oppervlak.
Er ontstaat een evenwicht tussen de oplossing en de oppervlaktelaag



Bij  er is een gebied waar
er is een gebied waar  hangt lineair af van de concentratie.
hangt lineair af van de concentratie.
G [mol/m2].
 -oppervlak dat wordt ingenomen door één mol van een stof
-oppervlak dat wordt ingenomen door één mol van een stof
Dan zal de tweedimensionale drukisotherm de vorm hebben 
 - tweedimensionale drukisotherm.
- tweedimensionale drukisotherm.
Verslaving  van S M:
van S M:

Bij  - de tweedimensionale druk neemt sterk toe. Bij
- de tweedimensionale druk neemt sterk toe. Bij  tweedimensionaal wordt vervormd, waardoor plotselinge groei ontstaat
tweedimensionaal wordt vervormd, waardoor plotselinge groei ontstaat  .
.
Een film die aan beide zijden door identieke fasen wordt begrensd, wordt dubbelzijdig genoemd. In dergelijke films wordt een constante beweging van de moederloog waargenomen.
Films die minder dan 5 nm dik zijn, worden zwarte films genoemd.

Adsorptielagen moeten twee kenmerken hebben: viscositeit en gemakkelijke mobiliteit, vloeibaarheid en elasticiteit.
Het Marangoni-effect is zelfgenezend.
Gibbs-driehoek,  - overdruk.
- overdruk.
De film is uitgerekt en doordat een deel van de vloeistof is verdwenen, stromen de oppervlakteactieve stoffen de vrije ruimte in. Gibbs-driehoek.
Het effect van de adsorptiesterkte van lichamen.
Er bevindt zich altijd een adsorptielaag op het oppervlak van de film, waarvoor dan
Langmuir-vergelijking:


 in tweedimensionale druk
in tweedimensionale druk
 - analoog van de Shishkovsky-vergelijking
- analoog van de Shishkovsky-vergelijking
Elektrokinetische verschijnselen. Elektrische dubbellaags (EDL).
Gelemholtz-model. Gouy-Chapman-theorie.
1808 Vlucht

U – gevormde buis, dompel er 2 elektroden in. De wet van communicerende vaten wordt overtreden en er vindt een verandering in het vloeistofniveau in de buis plaats - elektrokinetische verschijnselen.
Kinetische verschijnselen:
Elektroforese
Elektro-osmose
Stroom (stroom) potentieel
Sedimentatiepotentieel
1 en 2 ontstaan wanneer een potentiaalverschil wordt aangelegd; 3 en 4 veroorzaken ponsen en sedimentatie van colloïdale deeltjes de schijn van een potentiaalverschil.
Elektro-osmose is de beweging van een dispersiemedium ten opzichte van een stationaire verspreide fase onder invloed van een elektrische stroom.
Elektroforese – dit is de beweging van deeltjes in de verspreide fase ten opzichte van een stationair dispersiemedium onder invloed van een elektrische stroom.
P  De reden voor het optreden van elektrokinetische verschijnselen is de ruimtelijke scheiding van ladingen en het verschijnen van een dubbele elektrische laag.
De reden voor het optreden van elektrokinetische verschijnselen is de ruimtelijke scheiding van ladingen en het verschijnen van een dubbele elektrische laag.
De elektrische dubbellaag is een platte condensator, de ene plaat wordt gevormd door potentiaalbepalende ionen, de andere door tegenionen. De ionen worden op dezelfde manier verontreinigd als potentiaalbepalende co-ionen in het volume van de oplossing worden geduwd. Afstand tussen platen  . De potentiaal daalt lineair, het potentiaalverschil
. De potentiaal daalt lineair, het potentiaalverschil  .
.
Een extern potentiaalverschil veroorzaakt het optreden van een afschuifmodulus  is een paar krachten per oppervlakte-eenheid die langs het oppervlak van een vast lichaam werken.
is een paar krachten per oppervlakte-eenheid die langs het oppervlak van een vast lichaam werken.

In evenwicht is de afschuifmodulus gelijk aan de viskeuze wrijvingsmodulus (  ).
).

In onze voorwaarden  ,
,

 - Gelemholtz-Smalukowski-vergelijking
- Gelemholtz-Smalukowski-vergelijking
 - lineaire snelheid van faseverplaatsing.
- lineaire snelheid van faseverplaatsing.
E– elektrische veldsterkte.
 - potentiaalverschil tussen platen
- potentiaalverschil tussen platen
 - elektroforetische mobiliteit [m 2 /(V*s)].
- elektroforetische mobiliteit [m 2 /(V*s)].
Het Helemholtz-model houdt geen rekening met de thermische beweging van moleculen. In werkelijkheid is de verdeling van ionen in de dubbele laag complexer.
Gui en Chapman identificeerden de volgende oorzaken van DES:
De overgang van een ion van de ene fase naar de andere wanneer er een evenwicht is bereikt.
Ionisatie van vaste fase materie.
Voltooiing van het oppervlak met in het dispersiemedium aanwezige ionen.
Polarisatie van een externe stroombron.
De elektrische dubbellaag heeft een vage of diffuse structuur. De ionen hebben de neiging gelijkmatig over de diffuse laag verdeeld te zijn.
De diffuse laag bestaat uit counterinonen; de lengte van de laag wordt bepaald door hun kinetische energie. Bij temperaturen die het absolute nulpunt naderen, bevinden tegenionen zich zo dicht mogelijk bij potentiaalbepalende ionen.
Danya's theorie is gebaseerd op twee vergelijkingen:
Boltzmann-vergelijking

 - werken tegen de krachten van elektrostatische interactie.
- werken tegen de krachten van elektrostatische interactie.
 - volumetrische ladingsdichtheid.
- volumetrische ladingsdichtheid.

Poisson-vergelijking 
Omdat de dikte van de EDL veel kleiner is dan de deeltjesgrootte en voor een vlakke EDL de afgeleide met betrekking tot coördinaten  En
En  wordt afgeschaft.
wordt afgeschaft. 
Voor e y bij y<<1 функцию можно разложить в ряд Маклорена:

Laten we ons dan beperken tot twee termen van de reeks:


 - DEL-dikte is de afstand waarop de DEL-potentiaal afneemt e eenmaal.
- DEL-dikte is de afstand waarop de DEL-potentiaal afneemt e eenmaal.
Hoe lager de temperatuur, hoe minder  . Op T → 0 – vlak DEL. Hoe hoger de concentratie, hoe meer ik, hoe minder
. Op T → 0 – vlak DEL. Hoe hoger de concentratie, hoe meer ik, hoe minder  .
.





 “–” betekent dat de potentiaal afneemt met de afstand. =>
“–” betekent dat de potentiaal afneemt met de afstand. =>
=>

 ,
,
 - het potentieel neemt exponentieel af.
- het potentieel neemt exponentieel af.
Potentieel voor oppervlakteladingsdichtheid: 
Oppervlaktelading is een volumelading met het tegenovergestelde teken, geïntegreerd over de afstand.



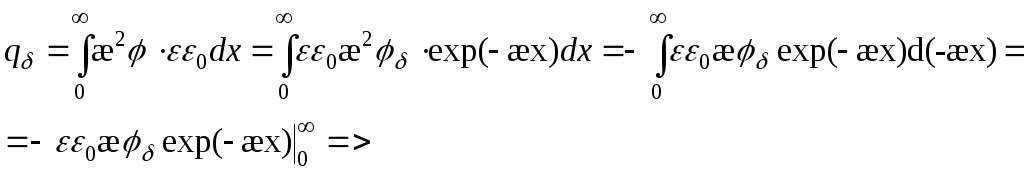
=>


Waar het potentieel 2,7 keer afneemt - 
Dubbellaagse capaciteit 
Het nadeel van de theorie is dat er geen rekening wordt gehouden met de aanwezigheid van de Helemholtz-laag, d.w.z. houdt geen rekening mee  , vandaar de fouten bij het bepalen van de belangrijkste parameters. Het verklaart ook niet de invloed van ionen van verschillende aard op de dikte van de elektrische dubbellaag.
, vandaar de fouten bij het bepalen van de belangrijkste parameters. Het verklaart ook niet de invloed van ionen van verschillende aard op de dikte van de elektrische dubbellaag.
Sterns theorie. Structuur van een colloïdale micel.
De elektrische dubbellaag bestaat uit twee delen: dicht en diffuus. Een dichte laag wordt gevormd als gevolg van de interactie van potentiaalvormende ionen met specifiek geadsorbeerde ionen. Deze ionen zijn in de regel gedeeltelijk of volledig gedehydrateerd en kunnen dezelfde of tegengestelde lading hebben als de potentiaalbepalende ionen. Het hangt af van de verhouding van de elektrostatische interactie-energie  en specifiek adsorptiepotentieel
en specifiek adsorptiepotentieel  . De dichte laagionen zijn gefixeerd. Het andere deel van de ionen bevindt zich in de diffuse laag; deze ionen zijn vrij en kunnen zich diep in de oplossing verplaatsen, d.w.z. van een gebied met een hogere concentratie naar een gebied met een lagere concentratie. De totale ladingsdichtheid bestaat uit twee delen.
. De dichte laagionen zijn gefixeerd. Het andere deel van de ionen bevindt zich in de diffuse laag; deze ionen zijn vrij en kunnen zich diep in de oplossing verplaatsen, d.w.z. van een gebied met een hogere concentratie naar een gebied met een lagere concentratie. De totale ladingsdichtheid bestaat uit twee delen.

 -lading van de Helmholtz-laag
-lading van de Helmholtz-laag
 -Diffuse laaglading
-Diffuse laaglading
Het oppervlak heeft een bepaald aantal adsorptiecentra, die elk een interactie aangaan met één tegenion. De constante van zo’n quasi-chemische reactie is gelijk aan:

 , Waar
, Waar  - molfractie van tegenionen in oplossing
- molfractie van tegenionen in oplossing
Helmholtz-distributie
De potentiaal neemt lineair af
Gouy potentiële distributie. Er is geen dichte laag, het potentieel neemt exponentieel af ten opzichte van de waarde 
Strenge distributie.
Aanvankelijk is de potentiële afname lineair en daarna exponentieel.
Wanneer bij elektroforese een elektrisch veld wordt aangelegd, beweegt niet het deeltje van de vaste fase direct, maar het deeltje van de vaste fase met een laag ionen eromheen. De DES herhaalt de vorm van het gedispergeerde fasedeeltje. Bij het aanleggen van een potentiaal wordt een deel van de diffuse laag afgescheurd. De breuklijn wordt genoemd glijdende grens.
De potentiaal die ontstaat bij de glijdende grens als gevolg van de scheiding van een deel van de diffuse laag, wordt genoemd elektrokinetisch potentieel(Zetapotentiaal  ).
).
Een gedispergeerd fasedeeltje met een omringende laag tegenionen en een dubbele elektrische laag wordt genoemd micel.
Regels voor het schrijven van colloïdale micellen:


1-1 opladen van elektrolyt
T – gedispergeerd fasedeeltje.
AA is de grens tussen de dichte en diffuse delen.
BB – glijdende grens.
De glijdende grens kan al dan niet samenvallen met lijn AA.
De pH-waarde waarbij de zeta-potentiaal nul is, wordt genoemd ISO-elektrisch punt.
CaCl 2 + Na 2 SO 4 → CaSO 4 ↓ + 2NaCl
1. Overtollig CaCl2
CaCl2 ↔ Ca2+ + 2Cl -
(CaSO 4 m∙nCa 2+ 2( n - x)Cl - ) 2 X + X Cl - - micelnotatie.
CaSO 4 m – aggregaat.
CaSO 4 m∙nCa 2+ – kern.
CaSO 4 m∙nCa 2+ 2( n - x)Cl - - deeltje.
2. Overtollig Na 2 SO 4
Na 2 SO 4 ↔2Na + + SO 4 2-
(CaSO 4 m∙nSO 4 2- 2(n-x)Na + ) 2x- 2xNa + - micel
CaSO 4 m – aggregaat.
CaSO 4 m∙nSO 4 2 + – kern.
CaSO 4 m∙nSO 4 2- 2(n-x)Na + - deeltje
Gelemholtz-Smoluchowski-vergelijking

 - lineaire snelheid van grensverplaatsing (bij elektro-osmose).
- lineaire snelheid van grensverplaatsing (bij elektro-osmose).
 - potentiaalverschil over de condensatorplaten (bij elektro-osmose).
- potentiaalverschil over de condensatorplaten (bij elektro-osmose).


 - volumetrische stroomsnelheid van de oplossing, S– dwarsdoorsnede van de cel.
- volumetrische stroomsnelheid van de oplossing, S– dwarsdoorsnede van de cel.

E– elektrische veldsterkte.


 (voor elektro-osmose).
(voor elektro-osmose).
Voor stromingspotentieel:

 - potentieel
- potentieel
 - druk op het membraan
- druk op het membraan
In de regel zijn de waarden van elektroforetische mobiliteiten en elektro-osmotische mobiliteiten minder dan berekende. Dit gebeurt vanwege:
Relaxatie-effect (wanneer een verspreid fasedeeltje beweegt, wordt de symmetrie van de ionische atmosfeer verbroken).
Elektroforetische remming (het optreden van extra wrijving als gevolg van de beweging van tegenionen).
Vervorming van stroomlijnen bij elektrisch geleidende deeltjes.
Verband tussen oppervlaktespanning en potentieel. Lippmann-vergelijking.
De vorming van EDL vindt spontaan plaats vanwege de wens van het systeem om zijn oppervlakte-energie te verminderen. In omstandigheden van constante T En P de algemene vergelijking van de eerste en tweede wet van de thermodynamica ziet er als volgt uit:
 (2)
(2)
(3), (1)=(3) =>
=>

 - 1e Lippmann-vergelijking.
- 1e Lippmann-vergelijking.
 - oppervlakteladingsdichtheid.
- oppervlakteladingsdichtheid.
 - differentiële capaciteit.
- differentiële capaciteit.
 - 2e Lippmann-vergelijking.
- 2e Lippmann-vergelijking.
MET- capaciteit.
Laten we de eerste Lippmann-vergelijking en de fundamentele adsorptievergelijking oplossen:
 ,
,


 , Dan
, Dan
 - Nernst-vergelijking
- Nernst-vergelijking
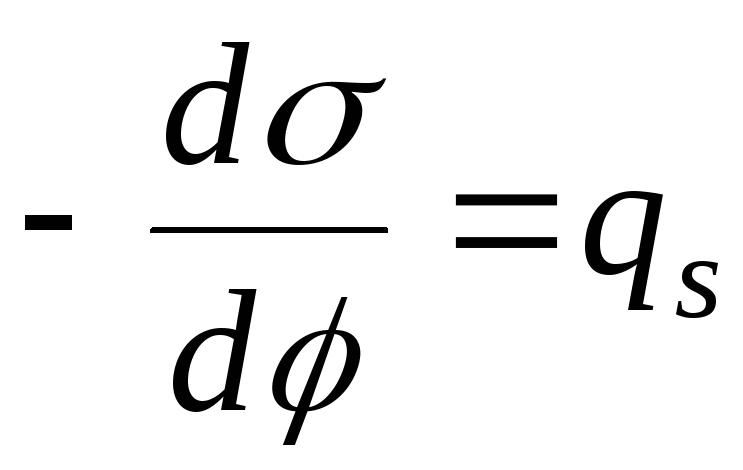 ,
,
 ,
,




 - vergelijking van de elektrocapillaire curve (ECC).
- vergelijking van de elektrocapillaire curve (ECC).
IN  :
: , Maar
, Maar 
Kationische oppervlakteactieve stoffen (CPAS) verminderen de kathodische tak van de EKC.
Anionische oppervlakteactieve stoffen (APS) verminderen de anodische tak van de EKC.
Niet-ionische oppervlakteactieve stoffen (NSAS) verminderen het middelste deel van de ECC.
Stabiliteit van verspreide systemen. Onsamenhangende druk.
Verspreide systemen kunnen worden onderverdeeld:
Systemen die thermodynamisch onstabiel zijn, kunnen kinetisch stabiel zijn vanwege de overgang naar een metastabiele toestand.
Er zijn twee soorten stabiliteit:
Sedimentatiestabiliteit (ten opzichte van de zwaartekracht).
Aggregatieve stabiliteit. (ten opzichte van hechting)
Coagulatie is een proces van deeltjesadhesie, wat leidt tot verlies van aggregatieve stabiliteit. Stolling kan worden veroorzaakt door temperatuurveranderingen, pH, roeren en ultrageluid.
Er wordt onderscheid gemaakt tussen coagulatie:
Omkeerbaar.
Onomkeerbaar.
Coagulatie vindt plaats met de introductie van elektrolyten.
Stollingsregels:

Film- dit is het deel van het systeem dat zich tussen twee grensvlakken bevindt.
Onsamenhangende druk treedt op wanneer de filmdikte scherp afneemt als gevolg van de interactie van naderende oppervlaktelagen.

“-” - naarmate de filmdikte afneemt, neemt de ontkoppelende druk toe.
P 0 is de druk in de bulkfase, die een voortzetting is van de tussenlaag.
P 1 – druk in de film.
Theorie van stabiliteit. DLFO (Deryagin, Landau, Fairway, Overbeck).
Volgens de DLFO-theorie bestaat ontkoppelingsdruk uit twee componenten:
Elektrostatisch PE (positief, het is te wijten aan de krachten van elektrostatische afstoting). Komt overeen met een afname van de Gibbs-energie bij toenemende filmdikte.
Moleculair P M (negatief, vanwege de werking van aantrekkelijke krachten). Het wordt veroorzaakt door filmcompressie als gevolg van chemische oppervlaktekrachten, de actieradius van krachten is tienden nm met een energie van ongeveer 400 kJ/mol.
Totale interactie-energie:

 - het systeem is aggregatief stabiel
- het systeem is aggregatief stabiel
 - onstabiel systeem
- onstabiel systeem
P  positieve component.
positieve component.
De toename is te wijten aan een toename van de potentiële energie wanneer dunne films worden gecomprimeerd. Voor films met een grote dikte wordt de overtollige ionenenergie gecompenseerd en is deze gelijk aan de energie-interactie in het volume van het dispersiemedium.
Als  (
( - film dikte,
- film dikte,  - ionenstraal) dunner worden van de film leidt tot het verdwijnen en verkleinen van moleculen en ionen met minimale oppervlakte-energie erin. Het aantal aangrenzende deeltjes neemt af, waardoor de potentiële energie van de in de film achterblijvende deeltjes toeneemt.
- ionenstraal) dunner worden van de film leidt tot het verdwijnen en verkleinen van moleculen en ionen met minimale oppervlakte-energie erin. Het aantal aangrenzende deeltjes neemt af, waardoor de potentiële energie van de in de film achterblijvende deeltjes toeneemt.


De DLVO-theorie beschouwt de interactie van deeltjes als de interactie van platen.

Deeltjes hebben geen interactie



 - Laplace-vergelijking,
- Laplace-vergelijking,  ,
,


Voor zwak geladen oppervlakken
Voor sterk geladen oppervlakken:

De moleculaire component is de interactie van twee atomen:
 ~
~
Interactie van een atoom met een oppervlak:

Laten we twee records nemen:
D  Om de moleculaire component te verkrijgen, is het noodzakelijk om alle interactie-energieën van de atomen van de rechter- en linkerplaat bij elkaar op te tellen.
Om de moleculaire component te verkrijgen, is het noodzakelijk om alle interactie-energieën van de atomen van de rechter- en linkerplaat bij elkaar op te tellen.
Waar  - Hamaker-constante (houdt rekening met de aard van op elkaar inwerkende lichamen).
- Hamaker-constante (houdt rekening met de aard van op elkaar inwerkende lichamen).
Dat. de interactie-energie van deeltjes in een systeem kan worden uitgedrukt met behulp van potentiële curven.
I – primair potentieel minimum. Dit is een zone van onomkeerbare coagulatie, de aantrekkingskrachten hebben de overhand.
II – zone van aggregatieve stabiliteit, afstotende krachten overheersen.
III – secundair potentieel minimum (of flocculatiezone). Er bevindt zich een elektrolytlaag tussen de deeltjes van de gedispergeerde fase, en de deeltjes kunnen worden gescheiden en overgebracht naar de zone van aggregatiestabiliteit.

Curve 1 – het systeem is aggregatief stabiel.
Curve 2 – stabiel in zone I, onstabiel in zone II.
Curve 3 – Er heeft coagulatie plaatsgevonden in het systeem.
Curve 4 – op punt 4 is de totale interactie-energie U=0,  , dit uiterste punt komt overeen met het begin van snelle coagulatie.
, dit uiterste punt komt overeen met het begin van snelle coagulatie.
Er zijn twee gevallen:
1. Licht geladen oppervlakken:
U = U E + U M = 0
 (1)
(1)
2)

 (2)
(2)



 - dit is de dikte van de laag die overeenkomt met het begin van het coagulatieproces.
- dit is de dikte van de laag die overeenkomt met het begin van het coagulatieproces.






 - voor zwak geladen oppervlakken
- voor zwak geladen oppervlakken
 Dan
Dan 
2. Voor sterk geladen oppervlakken:
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
Laten we vierkant maken (3)

Stolling:
Bij specifieke adsorptie kunnen ionen in super-equivalente hoeveelheden worden geadsorbeerd, zodat het oppervlak zijn lading kan veranderen. Het oppervlak wordt opgeladen.
Bij specifieke adsorptie kunnen niet alleen ionen met tegengesteld teken worden geadsorbeerd, maar ook ionen met hetzelfde teken.
Als ionen met hetzelfde teken als het oppervlak worden geadsorbeerd, zal er in de oppervlaktelaag geen daling van de potentiaal zijn, maar een toename ervan.

Neutralisatiecoagulatie (treedt op met de deelname van zwak geladen deeltjes en hangt niet alleen af van de lading van de elektrolyt-coagulator, maar ook van het potentieel aan de grens van de dichte en diffuse lagen).
Smoluchowski's theorie van snelle coagulatie.

Afhankelijkheid van de stollingssnelheid van de elektrolytconcentratie.
I – de stollingssnelheid is laag,
II – de stollingssnelheid is praktisch evenredig met de elektrolytconcentratie.
III – gebied van snelle coagulatie, de snelheid is vrijwel onafhankelijk van de concentratie.
Basisvoorzieningen:
De initiële sol is monodispers, soortgelijke deeltjes hebben een bolvorm.
Alle deeltjesbotsingen zijn effectief.
Wanneer twee primaire deeltjes botsen, ontstaat er een secundair deeltje. Secundair + primair = tertiair. Primair, secundair, tertiair – veelheid.
In termen van chemische kinetiek kan het coagulatieproces worden beschreven door de vergelijking:

De oplossing zal de vergelijking zijn: 
 - tijd van halve coagulatie. Dit is de tijd waarin het aantal soldeeltjes verdubbelt.
- tijd van halve coagulatie. Dit is de tijd waarin het aantal soldeeltjes verdubbelt.

 ,
,
 ,
,
 ,
,


Naarmate de multipliciteit toeneemt, verschuift het maximum van de coagulatiecurven naar grotere waarden  .
.
Gebreken:
Aanname van monodispersiteit.
Veronderstelling over de effectiviteit van alle botsingen.
Henry-adsorptie-isothermvergelijking
Als we het dynamische beeld van adsorptie in ogenschouw nemen, zal de waarde ervan groter zijn naarmate het aantal inslagen van gasmoleculen op het oppervlak groter is (d.w.z. hoe groter de gasdruk) en hoe langer de tijd dat het molecuul op het oppervlak blijft vanaf de moment van impact tot het moment van de overgang naar de gasfase. Daarom, maar De Boer, de omvang van de adsorptie
waarbij n het gemiddelde aantal moleculen is dat het oppervlak per tijdseenheid raakt, is τ de gemiddelde verblijftijd van moleculen op het oppervlak. Deze formule gaat ervan uit dat elke impact van een molecuul gepaard gaat met het vasthouden ervan aan het oppervlak, ongeacht of er al andere moleculen op zitten of niet. In werkelijkheid kan een molecuul dat een reeds bezette plek raakt, worden teruggekaatst in de gasfase of worden vastgehouden, maar de retentietijd zal anders zijn.
Het in aanmerking nemen van deze omstandigheden leidde tot de volgende formule:
Dit is de Henry-adsorptie-isothermvergelijking. Het betekent dat in een ideaal model de hoeveelheid adsorptie direct evenredig is met de druk van de damp of het gas. Deze afhankelijkheid kreeg deze naam naar analogie met de wet van Henry, bekend in de fysische chemie, volgens welke het volume gas opgelost in een vaste stof of vloeistof evenredig is aan de druk ervan. Volgens de geaccepteerde aannames zou de Henry-isotherm dus de experimentele gegevens moeten beschrijven die zijn verkregen bij kleine vullingen op homogene oppervlakken. De eerste veronderstelling is, zoals gezegd, gerechtvaardigd bij het bestuderen van adsorptie bij zeer lage drukken. Wat het tweede betreft, wordt adsorptie bijna altijd gemeten op heterogene oppervlakken. Adsorptie bij zeer lage drukken komt echter overeen met een zeer lage dekkingsgraad. Dit betekent dat alles afhangt van hoe heterogeen niet het hele oppervlak is, maar slechts een klein deel ervan, bedekt bij lage druk. Daarom zijn er in de literatuur voldoende voorbeelden van beide soorten te vinden. De constante K van de Henry-vergelijking (de raaklijn van de rechte lijn) hangt af van de temperatuur en energie van de adsorbaat-adsorbens-interactie, zoals blijkt uit vergelijking (4.4). Hoe lager de temperatuur en hoe groter de interactie van geadsorbeerde moleculen met het oppervlak van het adsorbens, hoe groter de K, hoe steiler de adsorptie-isotherm.
De veronderstelling dat moleculen met gelijke waarschijnlijkheid worden geadsorbeerd op welk oppervlak dan ook, inclusief de gebieden die al eerder bezet waren, is uiteraard een te ruwe veronderstelling, die alleen geschikt is voor een zeer kleine dekkingsgraad. Er kan nog een andere aanname worden gedaan, namelijk dat adsorptie alleen plaatsvindt op vrije delen van het oppervlak en dat het binnendringen van moleculen in reeds bezette gebieden niet tot adsorptie leidt. Deze aanname is gelijkwaardig aan het postulaat van monolaagadsorptie en wordt inderdaad vervuld, zoals we eerder zeiden, in het geval van chemische adsorptie, maar bij fysische adsorptie is de situatie ingewikkelder.
Een andere aanname bij het afleiden van de Henry-isothermvergelijking is de homogeniteit van het oppervlak, d.w.z. de gelijkwaardigheid van al zijn secties zullen we ongewijzigd laten. En ten slotte is de derde aanname in het nieuwe model dat wordt overwogen de afwezigheid van interactie tussen geadsorbeerde moleculen, d.w.z. We gaan ervan uit dat de tijd dat een molecuul op het oppervlak blijft, niet afhangt van waar het inslaat: in de onmiddellijke nabijheid van een ander molecuul of op grote afstand daarvan. Al deze aannames werden door Langmuir aanvaard toen hij in 1918 de adsorptie-isotherm afleidde.
De Langmuir-adsorptie-isothermvergelijking kan op verschillende manieren worden afgeleid. Langmuir heeft dit zelf afgeleid door de afhankelijkheid van de mate van adsorptie en desorptie te beschouwen van de mate van oppervlaktebedekking en door te overwegen dat bij evenwicht beide snelheden gelijk worden.
De thermodynamische afleiding van deze vergelijking werd gegeven door Volmer, en de statistische afleiding door Fowler.
In deze vorm is de Langmuir-vergelijking algemeen bekend. Het bevat twee constanten: en M kortweg monolaagcapaciteit genoemd (maximale adsorptie), en K is een constante die afhankelijk is van adsorptie-energie en temperatuur.
Adsorptie isotherm. Freundlich-vergelijking.
Adsorptiewaarde (absoluut A of overbodig G) wordt in elk specifiek geval bepaald door de temperatuur T en druk R(met gasvormig adsorbens) of temperatuur T en concentratie MET(tijdens adsorptie uit oplossingen). In de adsorptietheorie wordt bij het beschouwen van het adsorptie-evenwicht in de regel één van deze parameters constant gehouden. Dus, een vergelijking van de vorm A = f (p) T of G = f (c) T, die de hoeveelheid adsorptie relateert aan de druk of concentratie bij een constante temperatuur, wordt een adsorptie-isotherm genoemd. Adsorptie (als deze niet wordt uitgedrukt als een overschot, maar als een totaalgehalte) neemt altijd toe met toenemende evenwichtsdruk of concentratie. Omdat adsorptie dus een exotherm proces is bij toenemende temperatuur de waarde adsorptie neemt af. In afb. Figuur 26.9 toont de belangrijkste soorten adsorptie-evenwichtscurven. Adsorptie-isothermen bij drie temperaturen (T 1 > T 2 > T 3) komt overeen met afb. 26.9a.
Figuur 26.9. Adsorptie-evenwichtscurven: isothermen (a), isobaren (b) en isosteren (c) van adsorptie
Vergelijking die de grootte van adsorptie relateert aan de temperatuur bij constante evenwichtsdruk A = f(T) p of constante evenwichtsconcentratie G = f (T) s, wordt respectievelijk isobaren of isopycnalen van adsorptie genoemd (Fig. 26.9-b); Hier p 1 > p 2 > p 3 . Vergelijking van de vorm R= f (T) A, adsorptie-isostere (Fig. 26.9 -c), verbindt evenwichtsdruk met temperatuur bij een constante geadsorbeerde hoeveelheid; in dit geval EEN 1 > EEN 2 > EEN 3 .
De taak van elke adsorptietheorie is om er een wiskundige beschrijving van te maken op basis van een bepaald model van het adsorptieproces. Idealiter zou de vergelijking de afhankelijkheid van de evenwichtswaarde van adsorptie van de adsorbaatconcentratie in de bulkfase bij verschillende temperaturen moeten beschrijven, en ook de verandering in de adsorptiewarmte moeten voorspellen, afhankelijk van de vulling van het adsorbens. Meestal wordt de vergelijking voor de adsorptie-isotherm gevonden. De vorm van de adsorptie-isotherm op vaste stoffen hangt van veel parameters af: de eigenschappen van het adsorbens en het adsorbaat, de interactie van het adsorbens en het adsorbaat, de interactie van de adsorbaatmoleculen met elkaar in de gasfase en in de geadsorbeerde toestand. In het gebied van lage drukken (of concentraties) en overeenkomstige lage oppervlaktedekkingen is de interactie tussen adsorbaatmoleculen onbeduidend en de afhankelijkheid EEN = f(p) T reduceert tot de eenvoudigste vorm, genaamd Henry's wet:
A = kp of A = k"c(26.20)
Waar k En Naar"- adsorptiecoëfficiënt (of Henry's coëfficiënt), Met- concentratie van het adsorbens in de bulkfase, R- adsorberen dampdruk. Henry's coëfficiënt k is een maat voor de adsorptie-intensiteit. Er kan worden aangetoond dat elke theoretische isotherm, binnen de limiet (bij kleine vullingen), moet worden omgezet in de Henry-vergelijking.
In de buurt gemiddeld concentraties is de afhankelijkheid van de adsorptie van opgeloste stoffen van de concentratie empirisch goed beschreven Freundlich-vergelijking:
![]() (26.21)
(26.21)
Waar X- hoeveelheid geadsorbeerde stof, M- massa adsorbens, βi P - constanten die kenmerkend zijn voor elk adsorptiesysteem, met 0< 1/n< 1 . Volgens Freundllich is N is niet afhankelijk van vulling, hoewel deze verklaring niet helemaal juist is. Deze empirische vergelijking wordt vaak gebruikt voor geschatte adsorptieberekeningen. Meestal wordt het in logaritmische vorm gebruikt:
waardoor iemand een lineaire afhankelijkheid ln kan construeren A - ln c en grafisch beide constante parameters β en bepalen N.
Oppervlakteverschijnselen en adsorptie. Soorten adsorptie-interacties. Gasadsorptie-isothermen. Henry en Langmuir-vergelijking. Polymoleculaire adsorptie, BET-theorie.
OPPERVLAKTEFENOMENEN EN ADSORPTIE
Oppervlakte-energie. Adsorptie
Tot nu toe zijn de eigenschappen van heterogene systemen beschreven met behulp van parameters en toestandsfuncties die elk van de fasen als geheel karakteriseren. De eigenschappen van het deel van de fase dat aan het oppervlak grenst, verschillen echter van de eigenschappen van de fase in de bulk: in feite vormen de deeltjes die zich op het oppervlak van elke fase bevinden een speciale oppervlaktefase, waarvan de eigenschappen aanzienlijk verschillen van die van de fase. de eigenschappen van de interne gebieden van de fase. Deeltjes die zich op het oppervlak bevinden, bevinden zich in een andere omgeving dan deeltjes die zich in het volume van de fase bevinden, d.w.z. interageren zowel met homogene deeltjes als met deeltjes van een ander soort. Het gevolg hiervan is dat de gemiddelde energie g s van een deeltje dat zich op het fase-grensvlak bevindt, verschilt van de gemiddelde energie van hetzelfde deeltje in het volume van de fase g v (en de energie van een deeltje op het oppervlak kan groter of kleiner zijn dan de energie van een deeltje in het volume). Daarom is het belangrijkste kenmerk van de oppervlaktefase oppervlakte energie G s is het verschil tussen de gemiddelde energie van een deeltje dat zich op het oppervlak bevindt en een deeltje dat zich in het volume van de fase bevindt, vermenigvuldigd met het aantal deeltjes op het oppervlak N:
![]() (26.1)
(26.1)
Het is duidelijk dat de totale waarde van de oppervlakte-energie van een fase zal worden bepaald door de grootte van het oppervlak S. Daarom wordt het concept geïntroduceerd om het grensvlak te karakteriseren dat een bepaalde fase van een andere scheidt. oppervlaktespanningσ is de verhouding tussen oppervlakte-energie en het grensvlakgebied; de grootte van de oppervlaktespanning hangt alleen af van de aard van beide fasen. Net als de oppervlakte-energie van een fase kan oppervlaktespanning zowel positieve als negatieve waarden hebben. Oppervlaktespanning is positief als deeltjes op het oppervlak sterker interageren met deeltjes van dezelfde fase dan met deeltjes van een andere fase (en dus g s > g v). Volgens het principe van minimale vrije energie zal elke fase de neiging hebben om spontaan zijn oppervlakte-energie te verminderen; daarom heeft de fase, in het geval van een positieve oppervlaktespanning (σ > 0), de neiging zijn oppervlak te verkleinen. Als σ< 0, поверхностная энергия фазы будет уменьшаться при увеличении площади поверхности.
De invloed van de oppervlaktelaag van een fase op de algemene eigenschappen ervan wordt bepaald door het aandeel deeltjes dat zich op het oppervlak bevindt van het totale aantal deeltjes waaruit een bepaalde fase bestaat, d.w.z. de waarde van het specifieke oppervlak van de S/V-fase (oppervlakte per volume-eenheid). De vrije energie van fase G kan worden weergegeven als de som van oppervlakte-Gs- en volume-Gv-energieën, evenredig met respectievelijk het oppervlak en het volume van de fase:
Door deze uitdrukking te delen door het volume van de fase, verkrijgen we:
Uit vergelijking (IV.4) volgt dat voor dezelfde hoeveelheid fase (d.w.z. constant volume) de bijdrage van oppervlakte-energie aan de totale energie van de fase toeneemt met toenemend specifiek oppervlak, of, met andere woorden, mate van spreiding(fragmentatie) fasen. In het geval dat de mate van spreiding van de fase klein is (het specifieke oppervlak is onbeduidend), wordt de bijdrage van de oppervlakte-energie aan de totale energie van de fase gewoonlijk verwaarloosd. Bij het bestuderen wordt rekening gehouden met de bijdrage van de oppervlaktelaag aan de eigenschappen van de fase en het systeem als geheel verspreide systemen– heterogene systemen, waarvan een van de fasen continu is ( dispersie medium), en de andere is gefragmenteerd ( verspreide fase).
Op het grensvlak van een gecondenseerde (dat wil zeggen vaste of vloeibare) fase met een gas is de oppervlaktespanning altijd positief omdat de deeltjes van de gecondenseerde fase sterker met elkaar interageren dan met gasmoleculen. Volgens het principe van minimale vrije energie zal de gecondenseerde fase de neiging hebben om spontaan zijn oppervlakte-energie te verminderen. Dit kan het resultaat zijn van een afname van het oppervlak van de fase (daarom neemt een druppel vloeistof in gewichtloosheid de vorm aan van een bol), of een afname van de oppervlaktespanning wanneer nieuwe deeltjes op het fase-grensvlak verschijnen. - moleculen van een gas of een opgeloste stof. Het proces van spontane verandering in de concentratie van een stof op het grensvlak tussen twee fasen wordt genoemd adsorptie. Adsorbens is een stof op het oppervlak waarvan een verandering in de concentratie van een andere stof optreedt - adsorberen.
Adsorptie aan het grensvlak tussen oplossing en damp
In vloeibare oplossingen is de oppervlaktespanning σ een functie van de concentratie van de opgeloste stof. In afb. Figuur 4.1 presenteert drie mogelijke afhankelijkheid van de oppervlaktespanning van de oplossingsconcentratie (zogenaamde oppervlaktespanningisothermen). Stoffen waarvan de toevoeging aan een oplosmiddel de oppervlaktespanning vermindert, worden genoemd oppervlakteactieve stof(oppervlakteactieve stoffen), stoffen waarvan de toevoeging de oppervlaktespanning verhoogt of niet verandert – oppervlakte-inactief(PIAV).

Rijst. 26.1 Oppervlakte-isothermen Afb. 26.2 Adsorptie-isotherm
spanning van oppervlakteactieve oplossingen (1, 2) en PIAV. Oppervlakteactieve stof op het scheidingsvlak tussen oplossing en stoom
PIAV (3)
Een afname van de oppervlaktespanning en dus van de oppervlakte-energie treedt op als gevolg van de adsorptie van oppervlakteactieve stoffen aan het vloeistof-damp-grensvlak, d.w.z. het feit dat de concentratie oppervlakteactieve stof in de oppervlaktelaag van de oplossing groter is dan in de diepte van de oplossing.
Een kwantitatieve maatstaf voor adsorptie aan het grensvlak oplossing-damp is oppervlakte overtollig G (gamma), gelijk aan het aantal mol opgeloste stof in de oppervlaktelaag. De kwantitatieve relatie tussen de adsorptie (oppervlakteovermaat) van een opgeloste stof en de verandering in de oppervlaktespanning van de oplossing bij toenemende concentratie van de oplossing bepaalt Gibbs-adsorptie-isotherm:
De grafiek van de adsorptie-isotherm van oppervlakteactieve stoffen wordt getoond in Fig. 26.2. Uit vergelijking (26.5) volgt dat de richting van het proces - concentratie van de stof in de oppervlaktelaag of, omgekeerd, de aanwezigheid ervan in het volume van de vloeibare fase - wordt bepaald door het teken van de afgeleide dσ/dС. Een negatieve waarde van dit derivaat komt overeen met de accumulatie van een stof in de oppervlaktelaag (G > 0), een positieve waarde komt overeen met een lagere concentratie van een stof in de oppervlaktelaag vergeleken met de concentratie ervan in het grootste deel van de oplossing.
De waarde g = –dσ/dС wordt ook wel de oppervlakteactiviteit van de opgeloste stof genoemd. De oppervlakteactiviteit van een oppervlakteactieve stof bij een bepaalde concentratie C1 wordt grafisch bepaald door een raaklijn te tekenen aan de oppervlaktespanningsisotherm op het punt C = C1; in dit geval is de oppervlakteactiviteit numeriek gelijk aan de raaklijn van de hellingshoek van de raaklijn aan de concentratieas:
Het is gemakkelijk op te merken dat bij toenemende concentratie de oppervlakteactiviteit van de oppervlakteactieve stof afneemt. Daarom wordt de oppervlakteactiviteit van een stof gewoonlijk bepaald bij een oneindig kleine oplossingsconcentratie; in dit geval hangt de waarde ervan, aangegeven met go, alleen af van de aard van de oppervlakteactieve stof en het oplosmiddel. Terwijl ze de oppervlaktespanning van waterige oplossingen van organische stoffen bestudeerden, stelden Traube en Duclos de volgende empirische regel vast voor homologe reeksen oppervlakteactieve stoffen:
In elke homologe reeks bij lage concentraties verhoogt het verlengen van de koolstofketen met één CH 2 -groep de oppervlakteactiviteit met 3 tot 3,5 keer.
Voor waterige oplossingen van vetzuren wordt de afhankelijkheid van de oppervlaktespanning van de concentratie empirisch beschreven Shishkovsky-vergelijking:
Hier zijn b en K empirische constanten, en de waarde van b is hetzelfde voor de gehele homologe reeks, en de waarde van K neemt voor elk volgend lid van de reeks 3 tot 3,5 keer toe.

Rijst. 26.3 Beperk de oriëntatie van oppervlakteactieve moleculen in de oppervlaktelaag
De moleculen van de meeste oppervlakteactieve stoffen hebben een difiele structuur, d.w.z. bevatten zowel een polaire groep als een niet-polaire koolwaterstofradicaal. De rangschikking van dergelijke moleculen in de oppervlaktelaag is energetisch het gunstigst, op voorwaarde dat de polaire groep moleculen gericht is naar de polaire fase (polaire vloeistof), en de niet-polaire groep gericht is naar de niet-polaire fase (gas of niet-polaire vloeistof). polaire vloeistof). Bij lage oplossingsconcentraties verstoort thermische beweging de oriëntatie van oppervlakteactieve moleculen; naarmate de concentratie toeneemt, raakt de adsorptielaag verzadigd en wordt er een laag “verticaal” georiënteerde oppervlakteactieve moleculen gevormd op het grensvlak (Fig. 26.3). De vorming van een dergelijke monomoleculaire laag komt overeen met de minimale waarde van de oppervlaktespanning van de oppervlakteactieve oplossing en de maximale waarde van adsorptie G (Fig. 26.1-26.2); bij een verdere toename van de concentratie oppervlakteactieve stof in de oplossing veranderen de oppervlaktespanning en adsorptie niet.
Adsorptie aan het grensvlak tussen vast en gas
Bij de adsorptie van gassen aan vaste stoffen is het beschrijven van de interactie tussen adsorbaat- en adsorbensmoleculen een zeer complexe taak, omdat de aard van hun interactie, die de aard van adsorptie bepaalt, verschillend kan zijn. Daarom wordt het probleem meestal vereenvoudigd door twee extreme gevallen te beschouwen waarin adsorptie wordt veroorzaakt door fysische of chemische krachten: respectievelijk fysische en chemische adsorptie.
Fysische adsorptie ontstaat als gevolg van Van der Waals-interacties. Het wordt gekenmerkt door omkeerbaarheid en een afname van de adsorptie bij toenemende temperatuur, d.w.z. exothermiciteit, en het thermische effect van fysische adsorptie ligt gewoonlijk dicht bij de vloeibaarmakingswarmte van het adsorbaat (10 - 80 kJ/mol). Dit is bijvoorbeeld de adsorptie van inerte gassen op steenkool.
Chemische adsorptie(chemisorptie) wordt uitgevoerd door de chemische interactie van adsorbens- en adsorbaatmoleculen. Chemisorptie is gewoonlijk onomkeerbaar; chemische adsorptie is, in tegenstelling tot fysieke adsorptie, gelokaliseerd, d.w.z. adsorbaatmoleculen kunnen niet langs het oppervlak van het adsorbens bewegen. Omdat chemisorptie een chemisch proces is dat een activeringsenergie in de orde van grootte van 40-120 kJ/mol vereist, bevordert een temperatuurstijging het optreden ervan. Een voorbeeld van chemische adsorptie is de adsorptie van zuurstof aan wolfraam of zilver bij hoge temperaturen.
Benadrukt moet worden dat de verschijnselen van fysische en chemische adsorptie in zeer zeldzame gevallen duidelijk van elkaar te onderscheiden zijn. Meestal worden tussenliggende opties geïmplementeerd, wanneer het grootste deel van de geadsorbeerde stof relatief zwak gebonden is en slechts een klein deel sterk gebonden is. Zuurstof op metalen of waterstof op nikkel worden bij lage temperaturen bijvoorbeeld geadsorbeerd volgens de wetten van fysische adsorptie, maar naarmate de temperatuur stijgt, begint chemische adsorptie op te treden. Naarmate de temperatuur stijgt, begint de toename van de chemische adsorptie vanaf een bepaalde temperatuur de afname van de fysieke adsorptie te overlappen, daarom heeft de temperatuurafhankelijkheid van adsorptie in dit geval een duidelijk gedefinieerd minimum (Fig. 26.4).

Rijst. 26.4 Afhankelijkheid van het volume waterstof geadsorbeerd door nikkel van de temperatuur
Bij een constante temperatuur hangt de hoeveelheid geadsorbeerde stof alleen af van de evenwichtsdruk of concentratie van het adsorbaat; de vergelijking die deze grootheden in verband brengt, wordt de adsorptie-isotherm genoemd.
Adsorptie theorieën
Er bestaat geen uniforme theorie die alle soorten adsorptie op verschillende fasegrensvlakken redelijk correct zou beschrijven; Laten we daarom enkele van de meest voorkomende adsorptietheorieën bekijken die bepaalde soorten adsorptie beschrijven op het grensvlak van vast gas of vaste oplossing.
Langmuir's theorie van monomoleculaire adsorptie
De theorie van monomoleculaire adsorptie, ontwikkeld door de Amerikaanse chemicus I. Langmuir, is gebaseerd op de volgende principes.
1) Adsorptie is gelokaliseerd en wordt veroorzaakt door krachten die dicht bij chemische krachten liggen.
2) Adsorptie vindt niet plaats op het gehele oppervlak van het adsorbens, maar op actieve centra, dit zijn uitsteeksels of depressies op het oppervlak van het adsorbens, gekenmerkt door de aanwezigheid van de zogenaamde. vrije valenties. De actieve centra worden als onafhankelijk beschouwd (d.w.z. één actief centrum heeft geen invloed op de adsorptiecapaciteit van andere) en zijn identiek.
3) Elk actief centrum is alleen in staat tot interactie met één adsorbaatmolecuul; Als gevolg hiervan kan zich slechts één laag geadsorbeerde moleculen op het oppervlak vormen.
4) Het adsorptieproces is omkeerbaar en evenwicht– het geadsorbeerde molecuul wordt enige tijd vastgehouden door de actieve plaats, waarna het wordt gedesorbeerd; Zo ontstaat na enige tijd een dynamisch evenwicht tussen de processen van adsorptie en desorptie.

Rijst. 26.5 Monomoleculaire adsorptie-isotherm
Bij evenwicht is de adsorptiesnelheid gelijk aan de desorptiesnelheid. De desorptiesnelheid is recht evenredig met de fractie bezette actieve plaatsen (x), en de adsorptiesnelheid is recht evenredig met het product van de adsorbaatconcentratie en de fractie vrije actieve plaatsen (1 – x):
![]() (26.9)
(26.9)
Vanaf hier vinden we x:
Door de teller en de noemer van de rechterkant van vergelijking (26.10) te delen door k A, verkrijgen we:
 (26.11)
(26.11)
De maximaal mogelijke waarde van GO-adsorptie wordt bereikt op voorwaarde dat alle actieve centra bezet zijn door adsorbaatmoleculen, d.w.z. x = 1. Hieruit volgt dat x = Г/Го. Als we dit in vergelijking (26.11) invullen, krijgen we:
Vergelijking (26.13) geldt monomoleculaire adsorptie-isotherm, waarbij de adsorptiewaarde Г wordt verbonden met de adsorbaatconcentratie C. Hier is b een bepaalde constante waarde voor een gegeven adsorbens-adsorbaatpaar (de verhouding van de desorptie- en adsorptiesnelheidsconstanten), numeriek gelijk aan de adsorbaatconcentratie waarbij de helft van de actieve concentratie plaatsen zijn bezet. Schema Langmuir-adsorptie-isothermen getoond in afb. 26.5. De constante b kan grafisch worden bepaald door een raaklijn te tekenen aan de adsorptie-isotherm op punt C = 0.
Bij het beschrijven van het gasadsorptieproces in vergelijking (26.13) kan de concentratie worden vervangen door een proportionele waarde van de partiële gasdruk:
Langmuirs theorie van monomoleculaire adsorptie is toepasbaar om enkele processen van adsorptie van gassen en opgeloste stoffen bij lage drukken (concentraties) van het adsorbaat te beschrijven.
Polyani's theorie van polymoleculaire adsorptie
In de praktijk zijn er vaak (vooral bij dampadsorptie) zogenaamde S-vormige adsorptie-isothermen (Fig. 4.6), waarvan de vorm de mogelijke interactie van geadsorbeerde moleculen met het adsorbaat aangeeft, beginnend vanaf een bepaalde drukwaarde.

Rijst. 26.6 Polymoleculaire adsorptie-isotherm
Om dergelijke adsorptie-isothermen te beschrijven, stelde M. Polyani voor theorie van polymoleculaire adsorptie, gebaseerd op de volgende basisprincipes:
1. Adsorptie wordt puur veroorzaakt fysieke krachten.
2. Oppervlak van het adsorbens homogeen, d.w.z. er zijn geen actieve centra; adsorptiekrachten vormen een continu krachtveld nabij het oppervlak van het adsorbens.
3. Adsorptiekrachten werken op een afstand die groter is dan de grootte van het adsorbaatmolecuul. Met andere woorden: er zijn er enkele adsorptievolume, dat tijdens adsorptie wordt gevuld met adsorbaatmoleculen.
4. De aantrekking van een adsorbaatmolecuul door het oppervlak van het adsorbens is niet afhankelijk van de aanwezigheid van andere moleculen in het adsorptievolume, waardoor het mogelijk is polymoleculair adsorptie.
5. Adsorptiekrachten onafhankelijk van temperatuur en daarom verandert het adsorptievolume niet bij een verandering in temperatuur.
Freundlich-vergelijking
De theoretische concepten ontwikkeld door Langmuir en Polyany idealiseren en vereenvoudigen grotendeels het ware beeld van adsorptie. In feite is het oppervlak van het adsorbens heterogeen, is er interactie tussen de geadsorbeerde deeltjes, zijn de actieve centra niet volledig onafhankelijk van elkaar, enz. Dit alles compliceert de vorm van de isothermvergelijking. G. Freundlich toonde aan dat bij een constante temperatuur het aantal mol geadsorbeerd gas of opgeloste stof per massa-eenheid van het adsorbens (de zogenaamde specifieke adsorptie x/m) evenredig is aan de evenwichtsdruk (voor gas) of evenwichtsconcentratie (voor stoffen geadsorbeerd uit oplossing) van het adsorbens verhoogd tot een bepaalde kracht, die altijd kleiner is dan één:
Adsorptie aan het grensvlak van vaste oplossing
Moleculaire adsorptie uit oplossingen
De adsorptie-isothermen van opgeloste stoffen uit een oplossing lijken qua uiterlijk op de adsorptie-isothermen voor gassen; voor verdunde oplossingen worden deze isothermen goed beschreven door de Freundlich- of Langmuir-vergelijkingen, als we de evenwichtsconcentratie van de opgeloste stof in de oplossing daarin substitueren. Adsorptie uit oplossingen is echter een veel complexer fenomeen vergeleken met gasadsorptie, aangezien adsorptie van het oplosmiddel vaak gelijktijdig plaatsvindt met de adsorptie van de opgeloste stof.

Rijst. 26.8 Oriëntatie van oppervlakteactieve moleculen op het oppervlak van het adsorbens
De afhankelijkheid van adsorptie van de structuur van adsorbaatmoleculen is zeer complex en het is vrij moeilijk om enige regelmaat af te leiden. De moleculen van veel organische stoffen bestaan uit polaire (hydrofiele) en niet-polaire (hydrofobe) groepen, d.w.z. zijn oppervlakteactieve stoffen. Wanneer ze worden geadsorbeerd op een vast adsorbens, worden oppervlakteactieve moleculen op een zodanige manier op het oppervlak georiënteerd dat het polaire deel van het molecuul naar de polaire fase is gericht en het niet-polaire deel naar de niet-polaire fase is gericht. Tijdens de adsorptie van alifatische carbonzuren uit waterige oplossingen op een niet-polair adsorbens - actieve kool - worden de moleculen dus door koolwaterstofradicalen naar het adsorbens georiënteerd; tijdens adsorptie uit benzeen (een niet-polair oplosmiddel) op een polair adsorbens - silicagel - zal de oriëntatie van de zuurmoleculen worden omgekeerd (Fig. 4.8).
Adsorptie uit elektrolytoplossingen
Adsorptie uit waterige oplossingen van elektrolyten vindt in de regel zodanig plaats dat overwegend ionen van één type uit de oplossing aan het vaste adsorbens worden geadsorbeerd. Preferentiële adsorptie uit een oplossing van een anion of kation wordt bepaald door de aard van het adsorbens en de ionen. Het mechanisme van adsorptie van ionen uit elektrolytoplossingen kan verschillen; onderscheid maken tussen uitwisseling en specifieke adsorptie van ionen.
Wissel adsorptie uit is een proces van ionenuitwisseling tussen een oplossing en een vaste fase, waarbij de vaste fase ionen met een bepaald teken (kationen of anionen) uit de oplossing absorbeert en in plaats daarvan een equivalent aantal andere ionen met hetzelfde teken in de oplossing afgeeft. Uitwisselingsadsorptie is altijd specifiek, d.w.z. voor een bepaald adsorbens zijn slechts bepaalde ionen in staat tot uitwisseling; uitwisselingsadsorptie is meestal onomkeerbaar.
Bij specifieke adsorptie adsorptie van ionen van welk type dan ook op het oppervlak van de vaste fase gaat niet gepaard met het vrijkomen in de oplossing van een equivalent aantal andere ionen van hetzelfde teken; de vaste fase krijgt een elektrische lading. Dit leidt ertoe dat een equivalent aantal ionen met tegengestelde ladingen nabij het oppervlak worden gegroepeerd onder invloed van elektrostatische aantrekkingskrachten, d.w.z. er ontstaat een elektrische dubbellaag. De interactie van ladingen geconcentreerd op het oppervlak leidt tot een afname van de oppervlakte-energie van het systeem. Voor het geval van specifieke adsorptie van een elektrolyt formuleerden Peskov en Fayans de volgende empirische regel ( Peskov-Fajans heersen):
Op het oppervlak van een kristallijne vaste stof wordt een ion specifiek geadsorbeerd uit een elektrolytoplossing, die in staat is zijn kristalrooster te voltooien of een slecht oplosbare verbinding kan vormen met een van de ionen in het kristal.
De wet van Henry kan als volgt worden geformuleerd: wanneer het systeem wordt verdund (de druk neemt af), neigt de verdelingscoëfficiënt naar een constante waarde die gelijk is aan de Henry-verdelingsconstante. Wat betreft de grootte van adsorptie A, zal deze wet als volgt worden geschreven:
E  Deze vergelijkingen vertegenwoordigen adsorptie-isothermen van een stof bij lage concentraties. In overeenstemming hiermee kan de wet van Henry als volgt worden geformuleerd: de hoeveelheid adsorptie bij lage gasdrukken (concentraties van een stof in oplossing) is recht evenredig met de druk (concentratie).
Deze vergelijkingen vertegenwoordigen adsorptie-isothermen van een stof bij lage concentraties. In overeenstemming hiermee kan de wet van Henry als volgt worden geformuleerd: de hoeveelheid adsorptie bij lage gasdrukken (concentraties van een stof in oplossing) is recht evenredig met de druk (concentratie).
Afwijkingen van de wet van Henry, uitgedrukt door veranderingen in activiteitscoëfficiënten in fasen, stellen ons meestal niet in staat het verloop van isothermen bij toenemende concentratie te beschrijven en te voorspellen.
(druk) van het adsorbaat. Om een theoretische adsorptie-isotherm te verkrijgen die een breder scala aan concentraties beschrijft, is het noodzakelijk om ideeën over het adsorptiemechanisme en specifieke modellen te gebruiken.
Met een groot deel van de afwijkingen van de adsorbaatactiviteitscoëfficiënt in de oppervlaktelaag ten opzichte van de eenheid kan rekening worden gehouden met behulp van het concept van adsorptie als een quasi-chemische reactie tussen het adsorbaat en de adsorptiecentra van het adsorbensoppervlak. Dit is het hoofdidee van de adsorptietheorie van Langmuir. Dit standpunt wordt verduidelijkt door de volgende aannames:
1) adsorptie is gelokaliseerd (moleculen bewegen niet langs het oppervlak) op individuele adsorptiecentra, die elk een interactie aangaan met slechts één adsorbaatmolecuul; als resultaat wordt een monomoleculaire laag gevormd;
2) adsorptiecentra zijn energetisch equivalent - het oppervlak van het adsorbens is equipotentiaal;
3) geadsorbeerde moleculen hebben geen interactie met elkaar.
Lyofiele dispersiesystemen. Classificatie en algemene kenmerken van pauwinnen. Thermodynamica en mechanisme van micellisatie. Structuur van micellen van oppervlakteactieve stoffen in waterige en koolwaterstofmedia. Oplossen.
Alle verspreide systemen, afhankelijk van het mechanisme van hun vormingsproces volgens de classificatie van P. A. Rebinder, zijn verdeeld in lyofiel, die worden verkregen door spontane dispersie van een van de fasen (spontane vorming van een heterogeen, vrij verspreid systeem), en lyofoob, als gevolg van dispersie en condensatie met oververzadiging (geforceerde vorming van een heterogeen vrij-dispers systeem).
Als bij toenemende concentratie van een stof de oppervlaktespanning aan het grensvlak afneemt, wordt zo’n stof oppervlakteactieve stof genoemd. Voor dergelijke stoffen de oppervlakteactiviteit
De aanwezigheid van hydrofiele en oleofiele delen in oppervlakteactieve moleculen is een karakteristiek onderscheidend kenmerk van hun structuur. Op basis van hun vermogen om te dissociëren in waterige oplossingen worden oppervlakteactieve stoffen onderverdeeld in ionisch en niet-ionisch. Ionische oppervlakteactieve stoffen zijn op hun beurt onderverdeeld in anionisch, kationisch en amfolytisch (amfoteer).
1) Anionische oppervlakteactieve stoffen dissociëren in water en vormen een oppervlakteactief anion.
2) Kationische oppervlakteactieve stoffen dissociëren in water en vormen een oppervlakteactief kation.
3) Amfolytische oppervlakteactieve stoffen bevatten twee functionele groepen, waarvan er één zuur en de andere basisch is, bijvoorbeeld carboxyl- en aminegroepen. Afhankelijk van de pH van het medium vertonen amfolytische oppervlakteactieve stoffen anionische of kationische eigenschappen.
Alle oppervlakteactieve stoffen zijn, wat betreft hun gedrag in water, onderverdeeld in echt oplosbaar en colloïdaal.
Werkelijk oplosbare oppervlakteactieve stoffen in oplossing bevinden zich in een moleculair gedispergeerde toestand tot concentraties die overeenkomen met hun verzadigde oplossingen en de scheiding van het systeem in twee continue fasen.
Het belangrijkste onderscheidende kenmerk van colloïdale oppervlakteactieve stoffen is het vermogen om thermodynamisch stabiele (lyofiele) heterogene disperse systemen (associatieve of micellaire colloïden) te vormen. De belangrijkste eigenschappen van colloïdale oppervlakteactieve stoffen, die hun waardevolle eigenschappen en wijdverbreide toepassing bepalen, zijn onder meer een hoge oppervlakteactiviteit; het vermogen tot spontane micellisatie - de vorming van lyofiele colloïdale oplossingen bij een oppervlakteactieve stofconcentratie boven een bepaalde waarde die de kritische micelconcentratie (KKM) wordt genoemd; het vermogen om op te lossen - een sterke toename van de oplosbaarheid van stoffen in oplossingen van colloïdale oppervlakteactieve stoffen als gevolg van hun "opname" in de micel; hoog vermogen om verschillende verspreidingssystemen te stabiliseren.
Bij concentraties boven KKM assembleren oppervlakteactieve moleculen zich tot micellen (geassocieerd) en de oplossing verandert in een micellair (associatief) colloïdaal systeem.
Onder een oppervlakteactieve micel wordt verstaan een associatie van amfifiele moleculen, waarvan de lyofiele groepen naar het overeenkomstige oplosmiddel zijn gericht, en de lyofobe groepen met elkaar zijn verbonden en de kern van de micel vormen. Het aantal moleculen waaruit een micel bestaat, wordt het associatiegetal genoemd, en de totale som van de molecuulmassa's van de moleculen in de micel, of het product van de massa van de micel door het getal van Avogadro, wordt de micellaire massa genoemd. Een bepaalde oriëntatie van amfifiele oppervlakteactieve moleculen in een micel zorgt voor minimale grensvlakspanning op het micel-medium grensvlak.
P  Bij concentraties oppervlakteactieve stoffen in een waterige oplossing die iets hoger zijn dan KKM, worden volgens de ideeën van Hartley bolvormige micellen (Hartley-micellen) gevormd. Het binnenste deel van Hartley-micellen bestaat uit met elkaar verweven koolwaterstofradicalen, de polaire groepen van oppervlakteactieve moleculen zijn naar de waterfase gericht. De diameter van dergelijke micellen is gelijk aan tweemaal de lengte van de oppervlakteactieve moleculen. Het aantal moleculen in een micel groeit snel binnen een smal concentratiebereik, en bij een verdere toename van de concentratie verandert het praktisch niet, maar neemt het aantal micellen toe. Bolvormige micellen kunnen 20 tot 100 moleculen of meer bevatten.
Bij concentraties oppervlakteactieve stoffen in een waterige oplossing die iets hoger zijn dan KKM, worden volgens de ideeën van Hartley bolvormige micellen (Hartley-micellen) gevormd. Het binnenste deel van Hartley-micellen bestaat uit met elkaar verweven koolwaterstofradicalen, de polaire groepen van oppervlakteactieve moleculen zijn naar de waterfase gericht. De diameter van dergelijke micellen is gelijk aan tweemaal de lengte van de oppervlakteactieve moleculen. Het aantal moleculen in een micel groeit snel binnen een smal concentratiebereik, en bij een verdere toename van de concentratie verandert het praktisch niet, maar neemt het aantal micellen toe. Bolvormige micellen kunnen 20 tot 100 moleculen of meer bevatten.
Naarmate de concentratie oppervlakteactieve stoffen toeneemt, doorloopt het micellaire systeem een reeks evenwichtstoestanden die verschillen in associatieaantallen, groottes en vormen van micellen. Wanneer een bepaalde concentratie wordt bereikt, beginnen de bolvormige micellen met elkaar te interageren, wat bijdraagt aan hun vervorming. Micellen hebben de neiging een cilindrische, schijfvormige, staafvormige, lamellaire vorm aan te nemen.
Micellenvorming in niet-waterige media is meestal het gevolg van aantrekkingskrachten tussen de polaire groepen oppervlakteactieve stoffen en de interactie van koolwaterstofradicalen met oplosmiddelmoleculen. De resulterende omgekeerde micellen bevatten binnenin ongehydrateerde of gehydrateerde polaire groepen, omgeven door een laag koolwaterstofradicalen. Het aantal associaties (van 3 tot 40) is aanzienlijk minder dan voor waterige oplossingen van oppervlakteactieve stoffen. In de regel neemt het toe met toenemende koolwaterstofradicaal tot een bepaalde grens.
Het fenomeen van het oplossen van stoffen in micellen van oppervlakteactieve stoffen wordt solubilisatie genoemd. De methode voor het opnemen van solubilisaatmoleculen in micellen in waterige oplossingen hangt af van de aard van de stof. Niet-polaire koolwaterstoffen bevinden zich, wanneer ze in micellen worden opgenomen, in de koolwaterstofkernen van de micellen. Polaire organische stoffen (alcoholen, aminen, zuren) zijn ingebed in een micel tussen oppervlakteactieve moleculen, zodat hun polaire groepen naar water gericht zijn, en de lipofiele delen van de moleculen parallel aan de koolwaterstofradicalen van de oppervlakteactieve stof zijn georiënteerd. Een derde methode voor het opnemen van een solubilisaat in micellen is ook mogelijk, vooral kenmerkend voor niet-ionische oppervlakteactieve stoffen. Gesolubiliseerde moleculen, bijvoorbeeld fenol, dringen niet door in de micellen, maar worden op hun oppervlak gefixeerd, gelegen tussen willekeurig gebogen polyoxyethyleenketens.
Oplossen is een spontaan en omkeerbaar proces; Bij een gegeven concentratie oppervlakteactieve stof en temperatuur komt er een zeer duidelijke verzadiging van de oplossing met solubilisaat overeen. Als resultaat van het oplossen worden stabiele disperse systemen verkregen, vergelijkbaar met spontaan gevormde ultramicroheterogene emulsies.
Bepaal de oppervlakte- en totale (interne) energie van 4 g watermist die deeltjes bevat met een dispersie van 5 10 7 M -1 , T= 20ºC, σ = 72 mJ/m 2 ; Dσ/ dT= - 0,16 mJ/(m 2 ·NAAR); ρ = 1000 kg/m2 3 .

Examenkaart nr. 10
De theorie van polymoleculaire adsorptie van BET: uitgangspunten, afleiding van de isothermvergelijking en de analyse ervan. Lineaire vorm van de BET-vergelijking. Bepaling van het specifieke oppervlak van adsorbentia, katalysatoren en andere poreuze lichamen.
De Langmuir-vergelijking kan alleen worden gebruikt onder de voorwaarde dat de adsorptie van een stof gepaard gaat met de vorming van een monomoleculaire laag.
In de meeste gevallen compenseert de monomoleculaire adsorptielaag de overtollige oppervlakte-energie niet volledig en kan de invloed van oppervlaktekrachten zich uitbreiden naar de tweede, derde en volgende adsorptielagen, wat resulteert in polymoleculaire adsorptie.
MET  Een moderne vorm van de polymoleculaire adsorptievergelijking - de basisvergelijking van de gegeneraliseerde Langmuir-theorie - werd voorgesteld door Brunauer, Emmett en Teller.
Een moderne vorm van de polymoleculaire adsorptievergelijking - de basisvergelijking van de gegeneraliseerde Langmuir-theorie - werd voorgesteld door Brunauer, Emmett en Teller.
In deze theorie is een aanvullende aanname naast die welke de basis vormden voor de afleiding van de isothermvergelijking van Langmuir het idee van de vorming op het oppervlak van het adsorbens van "opeenvolgende complexen" van adsorptiecentra met één, twee, drie, etc. adsorberen moleculen. Vervolgens kan het adsorptieproces worden weergegeven in de vorm van opeenvolgende quasi-chemische reacties:
De evenwichtsconstanten van deze reacties zijn respectievelijk gelijk
Laten we aangeven:
Het totale aantal actieve centra op het adsorbens, of de capaciteit van de monolaag, zal gelijk zijn aan

Na een reeks berekeningen met behulp van de reekstheorie verkrijgen we uiteindelijk:

D  Deze relatie is de basisvergelijking van de gegeneraliseerde Langmuir-theorie en wordt de BET-polymoleculaire adsorptievergelijking genoemd.
Deze relatie is de basisvergelijking van de gegeneraliseerde Langmuir-theorie en wordt de BET-polymoleculaire adsorptievergelijking genoemd.
Bij het verwerken van experimentele resultaten wordt de BET-vergelijking meestal in lineaire vorm gebruikt:
Hiermee kunt u zowel de constante parameters A ∞ als C grafisch bepalen:
Door de experimentele bepaling van A ∞ kunnen we het specifieke oppervlak van het adsorbens berekenen (oppervlakte-eenheidsmassa van het adsorbens): ![]() .
.
Adsorptie- concentratie van een stof uit het volume van de fasen op het grensvlak daartussen. Adsorptie kan worden beschouwd als absorptie stof (adsorbaat) door het oppervlak van het adsorbens.
Adsorbens- een stof aan het oppervlak waarvan adsorptie plaatsvindt.
Adsorptief - een gas of opgeloste stof die kan adsorberen op het oppervlak van een adsorbens.
Adsorberen - geadsorbeerde substantie die zich op het oppervlak van het adsorbens bevindt. Vaak worden de begrippen “adsorptief” en “adsorbaat” geïdentificeerd
Onderscheiden fysieke adsorptie, optredend zonder chemische verandering in het adsorbaat en chemische adsorptie(chemisorptie), vergezeld van de chemische interactie van het adsorbens met het adsorbens.
Er vindt adsorptie plaats op fasegrenzen: vast - vloeistof, vast - gas, vloeistof - gas, vloeistof - vloeistof.
Als een stof wordt geadsorbeerd in de vorm van moleculen, spreken we van moleculair adsorptie, in de vorm van ionen - ionisch adsorptie.
Adsorptie is omkeerbaar, het omgekeerde proces wordt genoemd desorptie.
De snelheden van adsorptie en desorptie zijn aan elkaar gelijk adsorptie-evenwicht, wat overeenkomt met evenwichtsconcentratie adsorberen in oplossing of evenwichtsdruk in de gasfase.
Adsorptiewaarde(A) wordt gekenmerkt door de evenwichtshoeveelheid geabsorbeerde stof (X) per massa-eenheid vast adsorbens (m): [mol/kg of kg/kg]
Adsorptie isotherm- grafische weergave van de afhankelijkheid van de adsorptiewaarde van de evenwichtsconcentratie of evenwichtsdruk bij een gegeven constante temperatuur.
Er wordt onderscheid gemaakt tussen adsorptie monomoleculair, waarbij het adsorbaat het oppervlak van het adsorbens bedekt met een laag van één molecuul dik polymoleculair, waarbij adsorbaatmoleculen in meerdere lagen op het oppervlak van het adsorbens kunnen worden geplaatst.
Monomoleculaire adsorptie-isotherm heeft de vorm getoond in figuur 12 ( Langmuir-isotherm)
A Sectie I - antwoorden klein evenwichtsconcentraties (drukken), wanneer een klein deel van het oppervlak van het adsorbens wordt ingenomen door adsorbaatmoleculen, en de afhankelijkheid A - c (p) lineair is;
Sectie II - gemiddeld concentraties (drukken) waarbij een aanzienlijk deel van het adsorbensoppervlak wordt ingenomen door adsorbaatmoleculen;
c (p) Sectie III - waargenomen wanneer hoog evenwichtsconcentraties (drukken), wanneer het gehele oppervlak van het adsorbens wordt ingenomen door adsorbaatmoleculen en wordt bereikt grenswaarde van adsorptie (A).
Monomoleculaire adsorptie-isotherm is goed beschreven door de Langmuir-vergelijking:

Waar in een individuele constanten voor elke individuele stof bij adsorptie aan een specifiek adsorbens;
s, blz- evenwichtsconcentratie of evenwichtsdruk.
Bij lage evenwichtsconcentraties kan de waarde worden verwaarloosd Met of R in de noemer. Vervolgens wordt de Langmuir-vergelijking omgezet in de vergelijking van een rechte lijn die door de oorsprong gaat:
A = A in c of A = A op blz
Bij hoge evenwichtsconcentraties we kunnen de hoeveelheid in de noemer verwaarlozen V. Vervolgens wordt de Langmuir-vergelijking omgezet in de vergelijking van een rechte lijn, onafhankelijk van Met of R: EEN = EEN
Voor praktische berekeningen het is noodzakelijk om de constanten van de Langmuir-vergelijking A en te kennen V. Door de vergelijking om te zetten in de lineaire vorm van een rechte lijn die niet door de oorsprong van de coördinaten gaat: , kunt u een grafiek maken van de afhankelijkheid 1/A - 1/s (Fig. 13).
1/A Het segment OB is gelijk aan 1/A. Coëfficiënt V kan worden gevonden op basis van het feit dat V gelijk aan de concentratie waarbij de adsorptiewaarde de helft van de limiet bedraagt.

In de grafiek bepaalt interpolatie de segment-OD, overeenkomend met 2/A en gelijk aan 1/v. Dan b = 1/OD.
De Langmuir-vergelijking is afgeleid op basis van de theorie van monomoleculaire adsorptie, die het volgende heeft belangrijkste bepalingen:
adsorptie van moleculen vindt alleen plaats op adsorptiecentra (topjes van onregelmatigheden en smalle poriën);
elk adsorptiecentrum kan slechts één adsorbaatmolecuul bevatten;
het adsorptieproces is omkeerbaar; adsorptie-evenwicht is dynamisch van aard. Geadsorbeerde moleculen worden slechts gedurende een bepaalde tijd door adsorptiecentra vastgehouden, waarna de desorptie van deze moleculen en de adsorptie van hetzelfde aantal nieuwe moleculen plaatsvindt.
Naast de Langmuir-vergelijking wordt deze in de praktijk vaak gebruikt Freundlich-vergelijking:
A = KS 1/n of A = KR 1/n, waarbij K en 1/n empirische constanten zijn.
De vergelijking is geschikter om te beschrijven adsorptie op poreus of poederachtig adsorbentia in de omgeving gemiddelde concentraties (drukken).
De adsorptie-isotherm van Freundlich heeft geen horizontale rechte lijn en de adsorptie neemt toe met toenemende concentratie (druk) (Fig. 14).

Rijst. 14
Voor het vinden van de constanten van de Freundlich-vergelijking het wordt met behulp van logaritme omgezet in de vergelijking van een rechte lijn die niet door de oorsprong gaat: log A = log K + 1/n log C.
In overeenstemming hiermee heeft de grafiek van log A versus log C of (P), opgebouwd uit experimentele gegevens, de vorm getoond in Fig. 15. Door extrapolatie naar de ordinaat-as wordt een segment OB verkregen dat gelijk is aan log K. De raaklijn van de hellingshoek van de rechte lijn BN aan de abscis-as is gelijk aan 1/n (). tg =)
Polymoleculaire adsorptie- waargenomen tijdens adsorptie op poreuze of poedervormige adsorbentia (silicagel, actieve kool, poeders en tabletten met medicinale stoffen). In dit geval gaat de adsorptie door totdat een dichte monomoleculaire laag wordt gevormd, zoals weergegeven in Fig. 16.

Rijst. 16.

Deze adsorptie komt overeen met een isotherm van een ander type (Fig. 17), de zogenaamde " S - isotherm".
Capillaire condensatie- het fenomeen van dampvervloeiing in de poriën of capillairen van een vast adsorbens, het wordt waargenomen tijdens de absorptie van gemakkelijk vloeibaar te maken gassen of dampen (bijvoorbeeld water, benzeen, enz.) als gevolg van polymoleculaire adsorptie. Waarin polymoleculaire laag vertegenwoordigt de dunste vloeistoffilm, die het binnenoppervlak van de porie bedekt. De lagen van dergelijke vloeistof die op smalle plaatsen met elkaar versmelten, vormen concave menisci, waaronder Er ontstaat een verminderde stoomdruk. Daarbij poriën gas(damp)moleculen aantrekken en gevuld met vloeistof, gevormd tijdens condensatie.
Bij lekkage adsorptie gecompliceerd door capillaire condensatie, de isotherm die overeenkomt met het vullen van de poriën (1) valt niet samen met de isotherm (2) die overeenkomt met het legen ervan (Fig. 18). Op de isotherm wordt het gevormd condensatiehysteresislus. De processen van adsorptie en desorptie zijn niet hetzelfde.
