Henrys lov om adsorpsjon. Langmuir adsorpsjon isoterm ligning. Lyofile spredningssystemer. Klassifisering og generelle egenskaper ved pav. Termodynamikk og micelliseringsmekanisme. Strukturen til overflateaktive miceller i vandige og hydrokarbonmedier. solubilisering
Når det gjelder samspillet mellom to atomer:
U er interaksjonsenergien;
U = U + U TREKK.
 - Lennard-Jones ligning
, c, b, m = konst
- Lennard-Jones ligning
, c, b, m = konst
I tilfeller av interaksjon av atomer med en fast overflate, er det nødvendig å oppsummere alle interaksjoner.
x er avstanden til overflaten
r - virkningsradiusen til tiltrekningskreftene
dV - volum
n er antall overflatemolekyler
U ADS. er adsorpsjonsinteraksjonsenergien



Ved adsorpsjon forsterkes tiltrekningen. Og når det gjelder interaksjon av den ikke-polar-ikke-polare typen, er adsorpsjonen hovedsakelig lokalisert i fordypningene.
elektrostatisk interaksjon.
Polar adsorbent - ikke-polart adsorbat
Ikke-polar adsorbent - polart adsorbat
En polar adsorbent er et polart adsorbat.
M  adsorbatmolekylet er representert som en dipol, og adsorbenten som en leder, der adsorbatmolekylet induserer en dipol speilsymmetrisk i forhold til den gitte.
adsorbatmolekylet er representert som en dipol, og adsorbenten som en leder, der adsorbatmolekylet induserer en dipol speilsymmetrisk i forhold til den gitte.
X - avstand til midten
Når du samhandler, oppstår potensialet:
 ,
,
 er dipolmomentet.
er dipolmomentet.
Potensialet har en tendens til å ta på seg maksimalverdien, dvs. dipoler har en tendens til å orientere seg vinkelrett på overflaten.
Siden en økning i temperatur fremmer veksten av Brownsk bevegelse, fører det til en retardasjon av adsorpsjonsprosessen.
Ved elektrostatisk interaksjon er adsorbatet hovedsakelig lokalisert på fremspring.
Fundamental adsorpsjonsligning.
Ved adsorpsjon omfordeles komponenten, noe som betyr at det kjemiske potensialet endres. Adsorpsjonsprosessen kan betraktes som overgangen av overflateenergi til kjemisk energi.
Lagvolum = 0, deretter den generaliserte ligningen I og II for termodynamikkens lov:
T = const; (1) = (2) => 

For et to-komponent system:
,
 ,
,
 =>
=>
=>
 - Gibbs adsorpsjonsligning
.
- Gibbs adsorpsjonsligning
.
For tilfelle av adsorpsjon av TV. kropp - gass:, 
 ,
,

 - isoterm
- isoterm
 - isobar
- isobar
 - isopykne
- isopykne
 - isoster
- isoster
Isoterm, isopycne, isostere er relatert til hverandre.
Fordi adsorpsjonsfunksjon 



Henry isoterm Langmuir isoterm



Termodynamikk. Adsorpsjon.
For kondensert media:
 ,
,
 ,
,
 - integrert endring i Gibbs-energien
.
- integrert endring i Gibbs-energien
.

P-trykk over en buet overflate, P S-trykk over en flat overflate
 - adsorpsjonspotensial
- adsorpsjonspotensial
Differensiell endring i entrapi 
 , Г = konst
, Г = konst
- differensiell entropi endring
- differensiell entalpi av adsorpsjon
 - isosterisk adsorpsjonsvarme
- isosterisk adsorpsjonsvarme
 - kondensasjonsvarme
- kondensasjonsvarme
 - netto adsorpsjonsvarme
- netto adsorpsjonsvarme

 ,
,




Qa er den integrerte adsorpsjonsvarmen, 
Qra er den integrerte netto adsorpsjonsvarmen,

Henrys ligning
Studiet av adsorpsjon er hemmet av inhomogeniteten til overflaten, så de enkleste regelmessighetene oppnås for homogene overflater.
La oss vurdere samspillet mellom gasser og en fast overflate, når en gass går fra en likevektstilstand i volumet til en likevektstilstand på overflaten. Dette tilfellet er analogt med likevekten til gasser i et gravitasjonsfelt.
 ,
,
 ,
=>
,
=> -Henrys ligning
-Henrys ligning
 - fordelingskoeffisient
- fordelingskoeffisient
I adsorpsjonsprosessen skjer en endring i kjemiske potensialer.
For bulkfase: 
For overflategass: 
I en tilstand av likevekt  , dvs.
, dvs.

I Henry-ligningen er konstanten ikke avhengig av konsentrasjonen
Henry-ligningen er gyldig i området med lave trykk og konsentrasjoner. Når konsentrasjonen øker, er 2 typer avvik fra Henrys lov mulig:
1 - positive avvik, D avtar, A minker
2 - negative avvik, D - øker, A - øker.
Typen av avvik bestemmes av overvekten av en eller annen type adsorbent-adsorbat-interaksjon.
Med en sterk adhesiv interaksjon øker aktivitetskoeffisientene - et positivt avvik. Ved sammenhengende interaksjoner observeres negative avvik.
monomolekylær adsorpsjon.
Langmuir isoterm.
De enkleste regelmessighetene ble oppnådd i Henrys teori. Langmuir foreslo en teori der adsorpsjon anses som en kvasikjemisk reaksjon. Hvori:
Overflaten er energetisk jevn.
Adsorpsjon er lokalisert, hvert adsorpsjonssenter samhandler med ett adsorbatmolekyl.
Adsorbatmolekyler interagerer ikke med hverandre.
Adsorpsjon er monolag.

 - overflate,
- overflate,  - adsorbat,
- adsorbat,  - adsorpsjonskompleks.
- adsorpsjonskompleks.
 , deretter konsentrasjonen av adsorpsjonssteder:
, deretter konsentrasjonen av adsorpsjonssteder:  ,
, - begrenser adsorpsjon.
- begrenser adsorpsjon.
 , deretter reaksjonskonstanten:
, deretter reaksjonskonstanten: 

 - Langmuir-ligningen.
- Langmuir-ligningen.
Adsorpsjon versus konsentrasjon
1 )
)
 ,
,
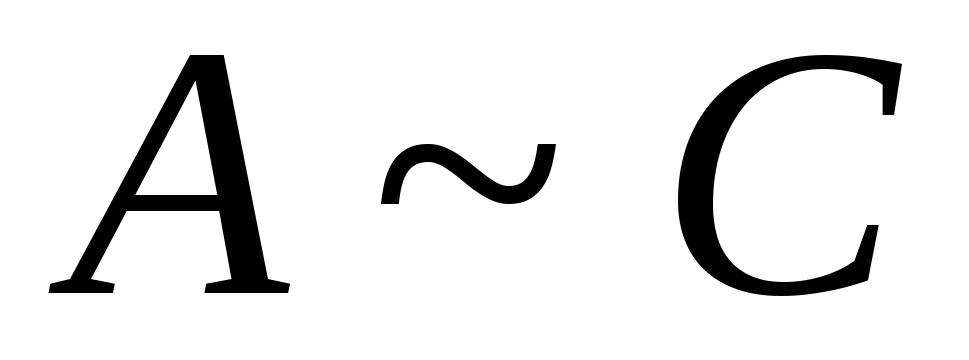
2) område med høye konsentrasjoner
 - begrensende adsorpsjon, dannelse av et monomolekylært lag
- begrensende adsorpsjon, dannelse av et monomolekylært lag
For Gibbs-energien: .
g er entropifaktoren.
Når det gjelder Henry-isotermen, karakteriserer Gibbs-energien overgangen til adsorbatet fra standardtilstanden i bulk til standardtilstanden på overflaten. I tilfellet med Langmuir-isotermen  karakteriserer graden av affinitet til adsorbenten og adsorbatet.
karakteriserer graden av affinitet til adsorbenten og adsorbatet.
 funnet fra van't Hoff isobar.
funnet fra van't Hoff isobar.

 , deretter
, deretter  , derfor
, derfor  .
.

 - grad av overflatefylling.
- grad av overflatefylling.




 - antall ledige stillinger,
- antall ledige stillinger,  - antall besatte plasser.
- antall besatte plasser.
 ,
,

De. i området med høye konsentrasjoner er antallet frie steder omvendt proporsjonalt med mengden adsorbat.
Adsorpsjon av en blanding av gasser på en homogen overflate.
I dette tilfellet betraktes adsorpsjonsprosessen som to parallelle reaksjoner.
 (1)
(1)

 (2)
(2)


Adsorpsjon av en blanding av gasser på en inhomogen overflate.
Ved ikke-homogen overflate bør man ikke begrense seg til middels utfyllinger.
Som et resultat av konkurranse er lokalisering av forskjellige adsorbater mulig på forskjellige typer steder.
I dette tilfellet forholdet  .
.
 ,
,
 er metningsdamptrykket til adsorbatet.
er metningsdamptrykket til adsorbatet.
 ,
,
 er adsorpsjonsvarmen.
er adsorpsjonsvarmen.
"+" - symmatisk avhengighet, "-" - antibatisk avhengighet, "H" - ingen korrelasjon.
"+" - adsorpsjon fortsetter i henhold til samme mekanisme. I de mest energimessig gunstige områdene adsorberes en gass med høy affinitet til overflaten hovedsakelig.
"-" - adsorpsjon foregår gjennom ulike mekanismer og opp til et visst tidspunkt er det ingen konkurranse om overflaten.
Monomolekylær adsorpsjon realiseres hovedsakelig under fysisk adsorpsjon av gasser ved lave verdier s, så vel som ved væske/gass-grensesnittet.
Polymolekylær adsorpsjon.
BET teori(Brunauer, Emmet, Teller).
I tilfellet når dannelsen av et monolag er utilstrekkelig til å kompensere for overflateenergien, er adsorpsjonen polymolekylær og kan betraktes som et resultat av tvungen kondensasjon under påvirkning av overflatekrefter.
Grunnleggende bestemmelser:
Når et adsorbatmolekyl treffer det okkuperte stedet, dannes et multippelsett.
Når du kommer nærmere s til s s antall gratis adsorpsjonssteder reduseres. Til å begynne med øker antall plasser besatt av singler, doubler osv. for deretter å reduseres. sett.
På s =s s adsorpsjon blir til kondens.
Det er ingen horisontale interaksjoner.
For det første laget utføres Langmuir-isotermen.
Overflaten betraktes som et sett med adsorpsjonssteder. Betingelsen for dynamisk likevekt er gyldig: kondenseringshastigheten på ledige steder er lik fordampningshastigheten fra okkuperte.
a er kondensasjonskoeffisienten (andelen av molekyler som kondenseres på overflaten);
 ,
,
Zm er maksimalt antall ledige plasser.
 - frekvens av vibrasjoner av atomer i retning vinkelrett på overflaten.
- frekvens av vibrasjoner av atomer i retning vinkelrett på overflaten.
For det første laget er de dynamiske likevektsforholdene:

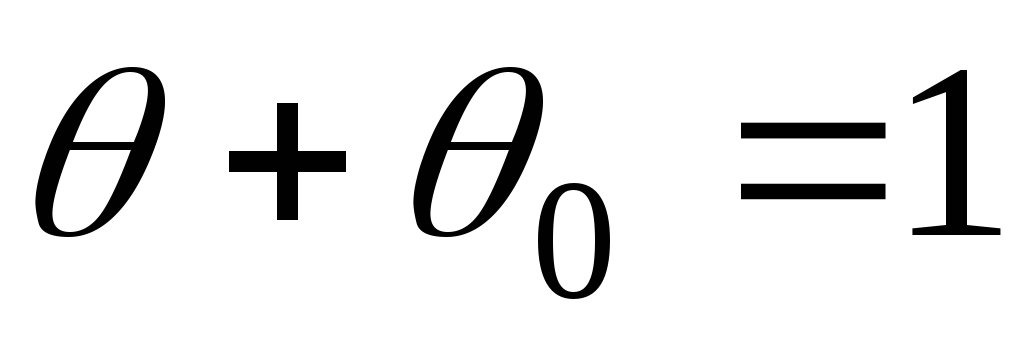

 , deretter
, deretter
 - Langmuir-ligningen.
- Langmuir-ligningen.
For det andre laget vil være sant: 
For det i-te laget: 
For enkelhets skyld antas det at a og ν er like for alle lag bortsett fra det første. For alle lag unntatt det første er adsorpsjonsvarmen konstant. For det siste laget er adsorpsjonsvarmen lik kondensasjonsvarmen. Som et resultat, ligningen
 (*)
(*)
C- konstant,
Når det gjelder BET-teori, konstanten Med karakteriserer Gibbs-energien til ren adsorpsjon. Ligningen inneholder bare en konstant, og denne ligningen er også veldig viktig for å bestemme det spesifikke overflatearealet til adsorbenten.
Siden varme frigjøres som et resultat av adsorpsjon, utføres bestemmelsen av spesifikke overflater ved lave temperaturer.
 ????????????
????????????


Teoriens hovedfeil– Forsømmelse av horisontale interaksjoner til fordel for vertikale.
Ligningen er i området  fra 0,05 til 0,3.
fra 0,05 til 0,3.
Hvor  <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 > 0,3 - interaksjonen av adsorbatet - adsorbat påvirker.
> 0,3 - interaksjonen av adsorbatet - adsorbat påvirker.
Redegjørelse for adsorbat-adsorbat interaksjoner.
Interaksjoner vises under adsorpsjon på en ikke-polar overflate av forgrenede molekyler eller molekyler. i stand til å danne assosiasjoner. I dette tilfellet endres formen på adsorpsjonsisotermene.
OG  sorbenten er ikke polar.
sorbenten er ikke polar.
Graf 1 tilsvarer svake interaksjoner adsorbat-adsorbat, sterk adsorbat-adsorbent.
Graf 2 tilsvarer en sterk adsorbat-adsorbat-interaksjon, en sterk adsorbat-adsorbent-interaksjon.
Graf 3 tilsvarer en sterk adsorbat-adsorbat-interaksjon, en svak adsorbat-adsorbent-interaksjon.
 ,
,

Ved interaksjon mellom adsorbatmolekyler er det nødvendig å ta hensyn til endringer i aktivitetskoeffisientene. Og denne ligningen er skrevet som:
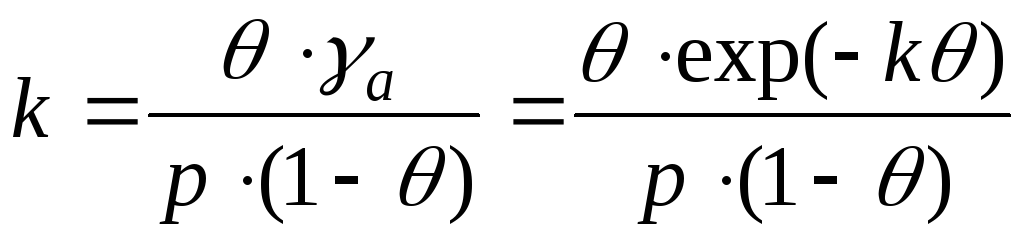 - ligningen til Frunkin, Fowler, Guggenheim.
- ligningen til Frunkin, Fowler, Guggenheim.
k er tiltrekningskonstanten.
Polans potensialteori.
Denne teorien utleder ikke noen form for adsorpsjonsisoterm, men gjør det mulig å beregne isotermer ved en annen temperatur.
Adsorpsjon er resultatet av tiltrekningen av adsorbatet til overflaten av adsorbenten på grunn av virkningen av adsorpsjonspotensialet, som ikke er avhengig av tilstedeværelsen av andre molekyler og avhenger av avstanden mellom overflaten og adsorbatmolekylet.
 ,
,
 - adsorpsjonspotensial.
- adsorpsjonspotensial.
Siden overflaten er inhomogen, erstattes avstanden med adsorpsjonsvolumet  .adsorpsjonsvolum er volumet innelukket mellom overflaten og punktet som tilsvarer den gitte verdien
.adsorpsjonsvolum er volumet innelukket mellom overflaten og punktet som tilsvarer den gitte verdien  .
.
Adsorpsjonspotensial er arbeidet med å overføre 1 mol av adsorbatet utenfor det gitte adsorpsjonsvolumet til et gitt punkt i adsorpsjonsvolumet (eller arbeidet med å overføre 1 mol mettet damp av adsorbatet, som er i likevekt med det flytende adsorbatet i fravær av adsorbenten, inn i dampfasen i likevekt med adsorbenten).

Karakteristisk kurve
 - adsorpsjonspotensial,
- adsorpsjonspotensial, 
For en gitt adsorbent og forskjellige adsorbater er følgende sant: 
For ulike typer adsorbater  ,
,
hvor  potensialer for adsorpsjonsisotermer ved relative trykk
potensialer for adsorpsjonsisotermer ved relative trykk  for adsorbat 1 og for adsorbat 2. Dette forholdet er en konstant verdi.
for adsorbat 1 og for adsorbat 2. Dette forholdet er en konstant verdi.
 - affinitetskoeffisient
- affinitetskoeffisient
Teori om kapillær kondensasjon.
Forløpet av adsorpsjonsprosessen avhenger i stor grad av strukturen til den porøse kroppen.
|
mikroporøs | |
|
Overgangsporøs | |
|
Makroporøs |
Når det gjelder mikroporøse sorbenter, overlapper feltene med adsorpsjonskrefter. Ved makroporøse sorbenter fungerer porene som transportkanaler. Kondensasjonsprosessene er mest betydningsfulle i forbigående porøse legemer. Kapillærkondensering starter ved visse verdier s og  når en del av overflateenergien allerede er kompensert. En nødvendig betingelse er at underlaget skal være selvbærende. Prosessen er beskrevet Thompson-Kelvin-ligningen.
når en del av overflateenergien allerede er kompensert. En nødvendig betingelse er at underlaget skal være selvbærende. Prosessen er beskrevet Thompson-Kelvin-ligningen.

 - for fukting er krumningssenteret i gassfasen.
- for fukting er krumningssenteret i gassfasen.
Ved kapillærkondensering har adsorpsjonsisotermen en hystereseform. Den nedre grenen tilsvarer adsorpsjonsprosessen, og den øvre grenen tilsvarer desorpsjonsprosessen.
Alle typer porer kan reduseres til tre typer:
|
konisk |
Sylindrisk med en lukket ende |
Sylindrisk med to åpne ender |
|
Prosessfylling utføres fra bunnen av poren. Adsorpsjonsisotermen og desorpsjonsisotermen faller i dette tilfellet sammen, siden adsorpsjonsprosessen begynner med en kule og desorpsjonsprosessen også begynner med at noen kuler forsvinner.
↓ |
Det er ingen hysterese. Forover og bakover slag er beskrevet av ligningen:
|
Det er ingen bunn noe sted, fyllingen av poren vil gå langs sylinderens vegger.
sylinder: Isoterm og vil ha en hystereseform.
↓ |


 - sfære,
- sfære, ,
,



PÅ  fuktingsforhold oppstår kondens ved lavere trykk, noe som er energetisk gunstig. Fra desorpsjonsgrenen oppnås porestørrelsesfordelingskurver.
fuktingsforhold oppstår kondens ved lavere trykk, noe som er energetisk gunstig. Fra desorpsjonsgrenen oppnås porestørrelsesfordelingskurver.
Differensialkurvens maksimum forskyves til venstre i forhold til integralets bøyningspunkt. Det totale volumet av små porer er lite, men har store overflatearealer. Når porestørrelsen øker, øker volumet deres som  , og området som
, og området som  , på grunn av dette observeres en forskyvning av maksimum av differensialkurven.
, på grunn av dette observeres en forskyvning av maksimum av differensialkurven.
Adsorpsjon ved fast-væske-grensesnittet.
Når det gjelder adsorpsjon ved fast-gass-grensesnittet, forsømte vi en komponent. Ved adsorpsjon ved faststoff-væske-grensesnittet, fortrenger adsorbatet løsemiddelmolekyler fra overflaten av adsorbenten.
 ,
,
Den riktige ligningen er:

 ,
,
N 1, N 2 - molfraksjoner av løsningsmidlet og komponenten, N 1 + N 2 \u003d 1, deretter
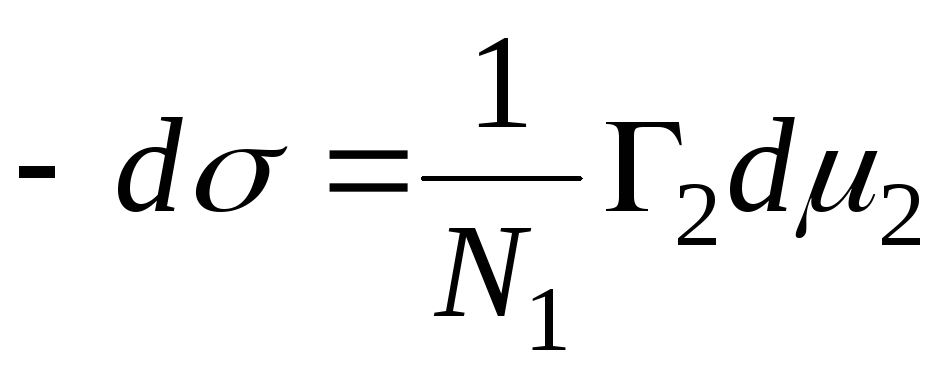 ,
=>
,
=>
 , deretter - adsorpsjonsligningen for fasegrensen faststoff - væske.
, deretter - adsorpsjonsligningen for fasegrensen faststoff - væske.
Adsorpsjon (G) > 0 ved  <
0
<
0
Hvis verdiene  for komponenten og løsningsmidlet er veldig forskjellige, i dette tilfellet avhengigheten G fra N har et ekstremum ved verdien N
~ 0,5.
for komponenten og løsningsmidlet er veldig forskjellige, i dette tilfellet avhengigheten G fra N har et ekstremum ved verdien N
~ 0,5.
E  hvis
hvis  har lignende verdier, i dette tilfellet kan tegnet på adsorpsjon endres. Avhengighet G fra N krysser x-aksen
har lignende verdier, i dette tilfellet kan tegnet på adsorpsjon endres. Avhengighet G fra N krysser x-aksen
Funksjon Kryss G(N) med abscisseaksen kalles adsorpsjonsazeotrop. Dette betyr at de to komponentene ikke kan separeres på denne adsorbenten.
Adsorpsjon isoterm ligning med utvekslingskonstant.
Under adsorpsjon ved faststoff-væske-grensesnittet blir komponentene konstant omfordelt mellom overflaten av adsorbenten og volumet av løsningen.
 - komponenter (- - referer til overflaten)
- komponenter (- - referer til overflaten)

 ,
,
 ,
, .
.
 ,
,


Adsorpsjon ved væske-gass-grensesnittet
R  La oss vurdere endringen i konsentrasjonsprofilen når væske-gass-grensesnittet krysses. La komponent 2 være flyktig.
La oss vurdere endringen i konsentrasjonsprofilen når væske-gass-grensesnittet krysses. La komponent 2 være flyktig.
Cs er konsentrasjonen i overflatelaget.
Basert på definisjonen av overflødig adsorpsjon

Hvis komponenten ikke er flyktig, vil adsorpsjonsverdien bli skrevet som følger:
P  ri
ri 

I ligningen  materiens natur er beskrevet av den deriverte
materiens natur er beskrevet av den deriverte  .
.
Overflatespenningsisotermen kan ha formen 1 eller 2:
1 - overflateaktive stoffer
2 - overflateaktive stoffer
Overflateaktivitet g er stoffenes evne til å redusere overflatespenningen i systemet.

 - tykkelse på overflatelaget
- tykkelse på overflatelaget
C s er konsentrasjonen av komponenten i overflatelaget
Med– volumkonsentrasjon
For en homolog serie er det en regel:
 - Traubeau Duclos regel
- Traubeau Duclos regel
For den homologe serien ser adsorpsjonsisotermen slik ut:
Vi skriver D i stedet for A, siden adsorpsjonen er for stor i overflatelaget.
Overflatespenningsisoterm:

 er overflatespenningen til det rene løsemidlet.
er overflatespenningen til det rene løsemidlet.
 - fundamental adsorpsjonsligning;
- fundamental adsorpsjonsligning;
 - Langmuir-ligningen.
- Langmuir-ligningen.
La oss løse dem sammen:

- Shishkovskys ligning.
B er en konstant for den homologe serien.
EN- når du flytter fra en homolog til en annen, øker den med 3-3,5 ganger 
![]()
1 - område med lave konsentrasjoner
![]()
2 - gjennomsnittlig konsentrasjon
3 - monomolekylært lag
Surfaktanter er amfifile molekyler, dvs. inkluderer en polar gruppe og et ikke-polart hydrokarbonradikal.
o er den polare delen av molekylet.
| er den ikke-polare delen av molekylet.
I et polart løsningsmiddel er overflateaktive molekyler orientert på en slik måte at den polare delen av molekylet vender mot løsningsmidlet, mens den upolare delen skyves inn i gassfasen.
I Shishkovsky-ligningen  , den er konstant for den homologe serien.
, den er konstant for den homologe serien.
Overflateaktiv handling begynner å vises med n>5. Ved konsentrasjoner høyere enn konsentrasjonen av det monomolekylære laget, skjer micellisering i overflateaktive løsninger.
Micelle- aggregatet av amfifile overflateaktive molekyler kalles, hvis hydrokarbonradikaler danner kjernen, og de polare gruppene omdannes til den vandige fasen.
Micelle masse - micellar masse.
H  antall molekyler er antall aggregering.
antall molekyler er antall aggregering.
Sfæriske miceller
Ved micellisering etableres en likevekt i løsningen
CMC er den kritiske micellekonsentrasjonen.
Siden vi anser micellen som en egen fase:
For den homologiske serien er det en empirisk ligning:
en er oppløsningsenergien til den funksjonelle gruppen.
b er adsorpsjonspotensialøkningen, arbeidet med adsorpsjon per metylenenhet.
er adsorpsjonspotensialøkningen, arbeidet med adsorpsjon per metylenenhet.
Tilstedeværelsen av en hydrokarbonkjerne i miceller gjør det mulig for forbindelser som er uløselige i vann å løse seg opp i vandige løsninger av overflateaktive stoffer, dette fenomenet kalles solubilisering (det som løses opp er et solubilisat, overflateaktivt middel er et solubiliseringsmiddel).
Slammet kan være helt upolart, kan inneholde både polare og ikke-polare deler, og vil være orientert som et overflateaktivt molekyl.
I alle fall, under solubilisering, oppstår en økning i micellær masse og aggregeringsnummer ikke bare på grunn av inkluderingen av solubilisatet, men også på grunn av en økning i antall overflateaktive molekyler som er nødvendige for å opprettholde likevektstilstanden.
Solubilisering er jo mer effektiv, jo lavere molekylvekt av solubilisatet.

 ~ 72 mN/m.
~ 72 mN/m.
 ~ 33 mN/m.
~ 33 mN/m.
Effektiviteten til overflateaktive midler avhenger av størrelsen på CMC.
2D overflatelagstrykk

→ -krefter av overflatespenning.
- todimensjonalt trykk.
Overflatelaget er en kraft lik forskjellen mellom overflatespenningene til en overflateaktiv løsning og et rent løsningsmiddel, rettet mot en ren overflate.
Det etableres en likevekt mellom løsningen og overflatelaget



På  det er et område hvor
det er et område hvor  lineært avhengig av konsentrasjon.
lineært avhengig av konsentrasjon.
G [mol/m 2].
 område okkupert av en mol av et stoff
område okkupert av en mol av et stoff
Da vil den todimensjonale trykkisotermen ha formen 
 er den todimensjonale trykkisotermen.
er den todimensjonale trykkisotermen.
Avhengighet  fra S M:
fra S M:

På  - todimensjonalt trykk øker kraftig. På
- todimensjonalt trykk øker kraftig. På  todimensjonal er deformert, noe som forårsaker en kraftig vekst
todimensjonal er deformert, noe som forårsaker en kraftig vekst  .
.
Filmen på begge sider begrenset av de samme fasene kalles dobbeltsidig. I slike filmer observeres en konstant bevegelse av moderluten.
Filmer mindre enn 5 nm tykke kalles svarte filmer.

Adsorpsjonslag bør ha to egenskaper: viskositet og lett bevegelighet, flyt og elastisitet.
Marangoni-effekten er selvhelbredende.
Gibbs Triangle,  - overtrykk.
- overtrykk.
Filmen strekkes, og på grunn av at en del av væsken er borte, skynder de overflateaktive stoffene inn i det ledige rommet. Gibbs trekant.
Effekt av adsorpsjonsstyrke til legemer.
Det er alltid et adsorpsjonslag på filmoverflaten, som da
Langmuir ligning:


 inn i todimensjonalt trykk
inn i todimensjonalt trykk
 - analog av Shishkovsky-ligningen
- analog av Shishkovsky-ligningen
elektrokinetiske fenomener. Dobbelt elektrisk lag (DES).
Helemholtz modell. Gouy-Chapman teori.
1808 Fly

U – formet rør, nedsenket i det 2 elektroder. Loven om kommuniserende fartøy brytes og det er en endring i væskenivået i røret - elektrokinetiske fenomener.
Kinetiske fenomener:
elektroforese
Elektroosmose
Strømnings (strømnings) potensial
Sedimentasjonspotensial
1 og 2 oppstår når en potensialforskjell påføres, 3 og 4 forårsaker stansing og sedimentering av kolloidale partikler utseendet til en potensialforskjell.
Elektroosmose er bevegelsen av et dispersjonsmedium i forhold til en stasjonær spredt fase under påvirkning av en elektrisk strøm.
elektroforese er bevegelsen av partikler av den dispergerte fasen i forhold til et stasjonært dispersjonsmedium under påvirkning av en elektrisk strøm.
P  Årsaken til forekomsten av elektrokinetiske fenomener er romlig separasjon av ladninger og utseendet til et dobbelt elektrisk lag.
Årsaken til forekomsten av elektrokinetiske fenomener er romlig separasjon av ladninger og utseendet til et dobbelt elektrisk lag.
Det elektriske dobbeltlaget er en flat kondensator, en plate er dannet av potensialbestemmende ioner, den andre av motinoer. Ionene blir også forurenset når potensialbestemmende koioner presses inn i hoveddelen av løsningen. Avstand mellom platene  . Potensialet faller lineært, potensialforskjellen
. Potensialet faller lineært, potensialforskjellen  .
.
En ekstern potensialforskjell forårsaker utseendet til en skjærmodul  er et par krefter per arealenhet som virker langs overflaten til et fast legeme.
er et par krefter per arealenhet som virker langs overflaten til et fast legeme.

Ved likevekt er skjærmodulen lik den viskøse friksjonsmodulen (  ).
).

I våre forhold  ,
,

 - Helemholtz-Smalukovsky-ligningen
- Helemholtz-Smalukovsky-ligningen
 - lineær hastighetsforskyvning i faser.
- lineær hastighetsforskyvning i faser.
E er den elektriske feltstyrken.
 - potensialforskjell mellom platene
- potensialforskjell mellom platene
 - elektroforetisk mobilitet [m 2 /(V * s)].
- elektroforetisk mobilitet [m 2 /(V * s)].
Helemholtz-modellen tar ikke hensyn til den termiske bevegelsen til molekyler. I virkeligheten er fordelingen av ioner i dobbeltlaget mer kompleks.
Gouy og Chapman identifiserte følgende årsaker til DES:
Overgangen til et ion fra en fase til en annen når likevekt er etablert.
Ionisering av fastfasestoff.
Komplettering av overflaten av ioner tilstede i dispersjonsmediet.
Polarisering fra en ekstern strømkilde.
Det elektriske dobbeltlaget har en uskarp eller diffus struktur. Ioner har en tendens til å være jevnt fordelt gjennom det diffuse laget.
Det diffuse laget består av motinnioner, lengden på laget bestemmes av deres kinetiske energi. Ved en temperatur som har en tendens til absolutt null, er motinoer så nærme som mulig potensialbestemmende ioner.
Denne teorien er basert på to ligninger:
Boltzmann-ligningen

 - arbeid mot kreftene til elektrostatisk interaksjon.
- arbeid mot kreftene til elektrostatisk interaksjon.
 er bulkladningstettheten.
er bulkladningstettheten.

Poisson-ligningen 
Siden DEL-tykkelsen er mye mindre enn partikkelstørrelsen, og for en flat DEL, er den deriverte med hensyn til koordinater  og
og  er opphevet.
er opphevet. 
For e y med y<<1 функцию можно разложить в ряд Маклорена:

Vi begrenser oss til to medlemmer av serien, da:


 - DEL-tykkelse er avstanden som DEL-potensialet avtar i e en gang.
- DEL-tykkelse er avstanden som DEL-potensialet avtar i e en gang.
Jo lavere temperatur, jo mindre  . Ved Т→0 – flat DES. Jo høyere konsentrasjon, jo mer jeg, jo mindre
. Ved Т→0 – flat DES. Jo høyere konsentrasjon, jo mer jeg, jo mindre  .
.





 "–" betyr at potensialet avtar med avstanden. =>
"–" betyr at potensialet avtar med avstanden. =>
=>

 ,
,
 - potensialet synker eksponentielt.
- potensialet synker eksponentielt.
Potensial for overflateladningstetthet: 
Overflateladning er en romladning med motsatt fortegn, integrert over avstand.



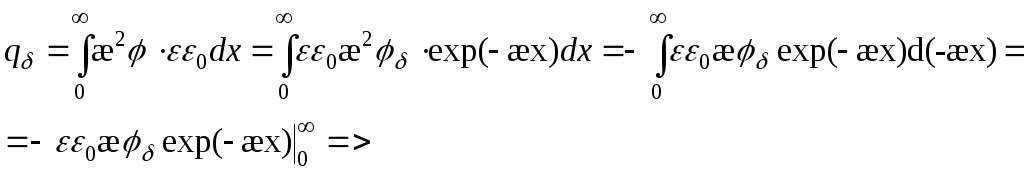
=>


Der potensialet reduseres med 2,7 ganger - 
Dobbeltlags kapasitet 
Ulempen med teorien er at tilstedeværelsen av Helemholtz-laget ikke tas i betraktning, d.v.s. tar ikke hensyn til  , derav feilene ved å bestemme hovedparametrene. Den forklarer heller ikke effekten av ioner av ulik natur på tykkelsen av det elektriske dobbeltlaget.
, derav feilene ved å bestemme hovedparametrene. Den forklarer heller ikke effekten av ioner av ulik natur på tykkelsen av det elektriske dobbeltlaget.
Sterns teori. Strukturen til en kolloidal micelle.
Det elektriske dobbeltlaget består av to deler: tett og diffust. Et tett lag dannes som et resultat av samspillet mellom potensialdannende ioner med spesifikt adsorberte. Disse ionene er som regel delvis eller fullstendig dehydrert og kan ha enten samme eller motsatt ladning til de potensialbestemmende ionene. Det avhenger av forholdet mellom energien til elektrostatisk interaksjon  og spesifikt adsorpsjonspotensial
og spesifikt adsorpsjonspotensial  . Ionene i det tette laget er fiksert. Den andre delen av ionene ligger i det diffuse laget, disse ionene er frie og kan bevege seg dypt inn i løsningen, dvs. fra et område med høyere konsentrasjon til et område med lavere konsentrasjon. Den totale ladningstettheten består av to deler.
. Ionene i det tette laget er fiksert. Den andre delen av ionene ligger i det diffuse laget, disse ionene er frie og kan bevege seg dypt inn i løsningen, dvs. fra et område med høyere konsentrasjon til et område med lavere konsentrasjon. Den totale ladningstettheten består av to deler.

 - Helmholtz lagladning
- Helmholtz lagladning
 -Diffus lagladning
-Diffus lagladning
Overflaten har et visst antall adsorpsjonssentre, som hver samhandler med ett motion. Konstanten for en slik kvasikjemisk reaksjon er:

 , hvor
, hvor  - molfraksjon av motioner i løsning
- molfraksjon av motioner i løsning
Helmholtz distribusjon
Potensialet avtar lineært
Gouy Potensial Distribusjon. Det er ikke noe tett lag, potensialet synker eksponentielt fra verdien 
Hekkfordeling.
Til å begynne med er potensialreduksjonen lineær, og deretter eksponentielt.
Når et elektrisk felt påføres ved elektroforese, er det ikke partikkelen i den faste fasen som beveger seg direkte, men partikkelen i den faste fasen med et lag av omgivende ioner. DES gjentar formen til partikkelen i den dispergerte fasen. Når et potensial påføres, rives en del av det diffuse laget av. Brytelinjen kalles glidende grense.
Potensialet som oppstår ved glidegrensen som følge av løsrivelse av en del av det diffuse laget kalles elektrokinetisk potensial(Zeta-potensial  ).
).
En partikkel av en dispergert fase, med et lag av motioner som omgir den og et dobbelt elektrisk lag, kalles micelle.
Regler for å skrive kolloidale miceller:


1-1 ladeelektrolytt
T er en partikkel av den dispergerte fasen.
AA er grensen mellom de tette og diffuse delene.
BB er glidegrensen.
Slippgrensen kan eller ikke falle sammen med linje AA.
pH-verdien der zetapotensialet er null kalles isoelektrisk punkt.
CaCl 2 + Na 2 SO 4 → CaSO 4 ↓ + 2 NaCl
1. I overkant av CaCl 2
CaCl 2 ↔ Ca 2+ + 2Cl -
(CaSO 4 m∙nCa 2+ 2( n-x)Cl - ) 2 x + x Cl - - rekordmiceller.
CaSO 4 m - tilslag.
CaSO 4 m∙nCa 2+ er kjernen.
CaSO 4 m∙nCa 2+ 2( n-x)Cl - partikkel.
2. I overkant av Na 2 SO 4
Na 2 SO 4 ↔2Na + + SO 4 2-
(CaSO 4 m∙nSO 4 2- 2(n-x)Na + ) 2x- 2xNa + - micelle
CaSO 4 m - tilslag.
CaSO 4 m∙nSO 4 2 + er kjernen.
CaSO 4 m∙nSO 4 2- 2(n-x)Na+-partikkel
Helemholtz-Smoluchowski-ligningen

 - lineær hastighet for forskyvning av grenser (i elektroosmose).
- lineær hastighet for forskyvning av grenser (i elektroosmose).
 - potensialforskjell på kondensatorplatene (ved elektroosmose).
- potensialforskjell på kondensatorplatene (ved elektroosmose).


 - volumetrisk strømningshastighet for løsningen, S er tverrsnittsarealet til cellen.
- volumetrisk strømningshastighet for løsningen, S er tverrsnittsarealet til cellen.

E er den elektriske feltstyrken.


 (for elektroosmose).
(for elektroosmose).
For strømningspotensialet:

 - potensiell
- potensiell
 - membrantrykk
- membrantrykk
Som regel er verdien av elektroforetiske mobiliteter og elektroosmotiske mobiliteter mindre enn de beregnede. Dette er på grunn av:
Avslappende effekt (under bevegelsen av en partikkel av den dispergerte fasen blir symmetrien til den ioniske atmosfæren krenket).
Elektroforetisk bremsing (forekomsten av ytterligere friksjon som følge av bevegelsen av motioner).
Forvrengning av strømlinjer ved elektrisk ledende partikler.
Sammenheng mellom overflatespenning og potensial. Lippmann ligning.
Dannelsen av DEL skjer spontant på grunn av systemets ønske om å redusere overflateenergien. I sammenheng med konstanthet T og s den generaliserte ligningen for termodynamikkens første og andre lov ser slik ut:
 (2)
(2)
(3), (1)=(3) =>
=>

 - 1. Lippmann-ligning.
- 1. Lippmann-ligning.
 er overflateladningstettheten.
er overflateladningstettheten.
 - differensiell kapasitans.
- differensiell kapasitans.
 - 2. Lippmann-ligning.
- 2. Lippmann-ligning.
Med- kapasitet.
Vi løser den første Lippmann-ligningen og den fundamentale adsorpsjonsligningen:
 ,
,


 , deretter
, deretter
 - Nernst-ligningen
- Nernst-ligningen
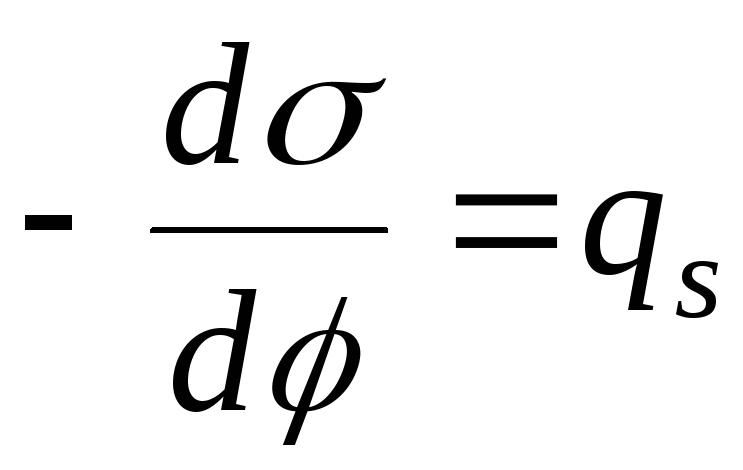 ,
,
 ,
,




 - ligningen for den elektrokapillære kurven (ECC).
- ligningen for den elektrokapillære kurven (ECC).
PÅ  :
: , men
, men 
Kationiske overflateaktive stoffer (CSAS) reduserer den katodiske grenen av ECC.
Anioniske overflateaktive stoffer (ASS) reduserer den anodiske grenen av ECC.
Ikke-ioniske overflateaktive stoffer (NSA) reduserer den midtre delen av ECC.
Stabilitet av disperse systemer. Kiletrykk.
Dispergerte systemer kan deles inn:
Termodynamisk ustabile systemer kan være kinetisk stabile på grunn av overgangen til en metastabil tilstand.
Det er to typer stabilitet:
Sedimentasjonsstabilitet (med hensyn til gravitasjon).
Aggregativ stabilitet. (i forhold til å stikke)
Koagulasjon er prosessen med at partikler holder seg sammen, noe som fører til tap av aggregatstabilitet. Koagulasjon kan være forårsaket av endringer i temperatur, pH, omrøring, ultralyd.
Skill koagulasjon:
Vendbar.
Irreversibel.
Koagulering fortsetter med innføring av elektrolytter.
Koagulasjonsregler:

Film– Dette er den delen av systemet som ligger mellom to grensesnitt.
usammenhengende press oppstår med en kraftig reduksjon i filmtykkelsen som følge av samspillet mellom overflatelag som nærmer seg.

«-» - når filmtykkelsen avtar, øker det usammenhengende trykket.
P 0 er trykket i bulkfasen, som er en fortsettelse av mellomsjiktet.
P 1 er trykket i filmen.
Teori om stabilitet. DLFO (Deryagin, Landau, Fairway, Overbeck).
I følge DLVO-teorien skilles to komponenter i det usammenhengende trykket:
elektrostatisk PE (positiv, det er på grunn av kreftene til elektrostatisk frastøtning). Tilsvarer en reduksjon i Gibbs-energien med økende filmtykkelse.
Molekylær PM (negativ, på grunn av virkningen av attraktive krefter). Det er forårsaket av komprimering av filmen på grunn av kjemiske overflatekrefter, kreftenes virkningsradius er tideler av en nm med en energi i størrelsesorden 400 kJ/mol.
Total interaksjonsenergi:

 - systemet er aggregert stabilt
- systemet er aggregert stabilt
 - ustabilt system
- ustabilt system
P  positiv komponent.
positiv komponent.
Økningen skyldes økningen i potensiell energi under komprimering av tynne filmer. For tykke filmer kompenseres overskuddet av ioneenergi og er lik energiinteraksjonen i hoveddelen av dispersjonsmediet.
Hvis  (
( - filmtykkelse,
- filmtykkelse,  - radius av ion) tynning av filmen fører til forsvinning og reduksjon av molekyler og ioner i den med et minimum av overflateenergi. Antallet nabopartikler avtar, som et resultat av at den potensielle energien til partiklene som er igjen i filmen øker.
- radius av ion) tynning av filmen fører til forsvinning og reduksjon av molekyler og ioner i den med et minimum av overflateenergi. Antallet nabopartikler avtar, som et resultat av at den potensielle energien til partiklene som er igjen i filmen øker.


DLVO-teorien anser interaksjonen mellom partikler som interaksjonen mellom plater.

Partikler samhandler ikke



 - Laplace-ligningen,
- Laplace-ligningen,  ,
,


For svakt ladede overflater
For svært ladede overflater:

Den molekylære komponenten er interaksjonen mellom to atomer:
 ~
~
Interaksjon mellom et atom og en overflate:

La oss ta to poster:
D  For å oppnå den molekylære komponenten, er det nødvendig å summere alle interaksjonsenergiene til atomene på høyre og venstre plate.
For å oppnå den molekylære komponenten, er det nødvendig å summere alle interaksjonsenergiene til atomene på høyre og venstre plate.
hvor  - Hamakers konstant (tar hensyn til naturen til samvirkende kropper).
- Hamakers konstant (tar hensyn til naturen til samvirkende kropper).
At. interaksjonsenergien til partikler i et system kan uttrykkes ved hjelp av potensielle kurver.
I er det primære potensielle minimumet. Dette er en sone med irreversibel koagulasjon, tiltrekningskreftene råder.
II - sone med aggregativ stabilitet, frastøtende krefter råder.
III - sekundært potensial minimum (eller flokkuleringssone). Mellom partiklene i den dispergerte fasen er det et elektrolyttlag, og partiklene kan separeres og overføres til sonen med aggregeringsstabilitet.

Kurve 1 – systemet er samlet sett stabilt.
Kurve 2 er stabil i sone I, ikke stabil i sone II.
Kurve 3 - koagulering skjedde i systemet.
Kurve 4 - ved punkt 4, den totale energien for interaksjon U=0,  , tilsvarer dette ekstremumpunktet begynnelsen av rask koagulasjon.
, tilsvarer dette ekstremumpunktet begynnelsen av rask koagulasjon.
Det er to tilfeller:
1. Overflater er svakt ladet:
U \u003d U E + U M \u003d 0
 (1)
(1)
2)

 (2)
(2)



 - dette er tykkelsen på laget som tilsvarer begynnelsen av koagulasjonsprosessen.
- dette er tykkelsen på laget som tilsvarer begynnelsen av koagulasjonsprosessen.






 - for svakt ladede overflater
- for svakt ladede overflater
 deretter
deretter 
2. For svært ladede overflater:
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
La oss kvadre (3)

Koagulasjon:
Ved spesifikk adsorpsjon kan ioner adsorberes i en superekvivalent mengde på en slik måte at overflaten kan endre ladningen. Overflaten lades opp.
Ved spesifikk adsorpsjon kan ikke bare ioner med motsatte fortegn, men også av ett ion adsorberes.
Hvis ioner av samme tegn som overflaten blir adsorbert, vil det i overflatelaget ikke være en dråpe i potensialet, men dets vekst.

Nøytraliseringskoagulering (oppstår med deltakelse av svakt ladede partikler og avhenger ikke bare av ladningen til den koagulerende elektrolytten, men også av potensialet ved grensen til de tette og diffuse lagene).
Smoluchowskis teori om rask koagulasjon.

Avhengighet av koagulasjonshastighet på elektrolyttkonsentrasjon.
I – koagulasjonshastigheten er lav,
II - koagulasjonshastigheten er praktisk talt proporsjonal med elektrolyttkonsentrasjonen.
III - området med rask koagulasjon, hastigheten er praktisk talt uavhengig av konsentrasjon.
Grunnleggende bestemmelser:
Den opprinnelige solen er monodispers, lignende partikler har en sfærisk form.
Alle partikkelkollisjoner er effektive.
Når to primærpartikler kolliderer, dannes det en sekundærpartikkel. Sekundær + primær = tertiær. Primær, sekundær, tertiær - multiplisitet.
Når det gjelder kjemisk kinetikk, kan koagulasjonsprosessen beskrives med ligningen:

Løsningen vil være ligningen: 
 - tid for halv koagulasjon. Dette er tiden hvor antall solpartikler reduseres med 2 ganger.
- tid for halv koagulasjon. Dette er tiden hvor antall solpartikler reduseres med 2 ganger.

 ,
,
 ,
,
 ,
,


Når multiplisiteten øker, skifter maksimum av koagulasjonskurvene mot større verdier  .
.
Feil:
Antagelse om monodispersitet.
Antakelsen om effektiviteten av alle kollisjoner.
Henry adsorpsjon isoterm ligning
Hvis vi tar i betraktning det dynamiske bildet av adsorpsjon, vil verdien være jo større, jo større antall påvirkninger av gassmolekyler på overflaten (dvs. jo større gasstrykk) og jo lenger tid molekylet forblir på overflaten fra kollisjonsøyeblikket til øyeblikket det går tilbake til gassfasen. Derfor, men de Boer, adsorpsjonsverdien
hvor n er gjennomsnittlig antall molekyler som treffer overflaten per tidsenhet, τ er gjennomsnittlig oppholdstid for molekylene på overflaten. Denne formelen antar at hver påvirkning av et molekyl er ledsaget av dens forsinkelse på overflaten, uavhengig av om det allerede er andre molekyler på det eller ikke. I virkeligheten kan et molekyl som treffer et allerede okkupert sted bli reflektert tilbake i gassfasen eller forsinket, men retensjonstiden vil være annerledes.
Regnskap for disse omstendighetene førte til følgende formel:
Dette er Henrys adsorpsjonsisotermligning. Det betyr at i den ideelle modellen er mengden av adsorpsjon direkte proporsjonal med damp- eller gasstrykket. Denne avhengigheten mottok dette navnet i analogi med Henrys lov kjent i fysisk kjemi, ifølge hvilken volumet av en gass oppløst i et fast stoff eller væske er proporsjonalt med trykket. Så, i henhold til de aksepterte forutsetningene, bør Henry-isotermen beskrive de eksperimentelle dataene oppnådd ved lave fyllinger på homogene overflater. Den første antagelsen er som sagt begrunnet i studiet av adsorpsjon ved svært lave trykk. Når det gjelder det andre, måles adsorpsjon nesten alltid på inhomogene overflater. Adsorpsjon ved svært lave trykk tilsvarer imidlertid svært lave dekningsgrader. Dette betyr at alt avhenger av hvor inhomogen ikke hele overflaten er, men bare en liten brøkdel av den, som dekkes ved lavt trykk. Derfor kan man i litteraturen finne nok eksempler av begge slag. Konstanten K til Henry-ligningen (tangensen til helningen til den rette linjen) avhenger av temperaturen og interaksjonsenergien til adsorbatet - adsorbenten, som man kan se av ligning (4.4). Jo lavere temperatur og jo større interaksjon mellom adsorberte molekyler og overflaten av adsorbenten er, jo større K, jo brattere er adsorpsjonsisotermen.
Selvfølgelig er antagelsen om at molekyler adsorberes med samme sannsynlighet på en hvilken som helst del av overflaten, inkludert de som allerede er okkupert tidligere, en for grov antakelse, egnet bare for svært små dekningsgrader. En annen antakelse kan gjøres, som er at adsorpsjon kun skjer på frie områder av overflaten og at ethvert treff av molekyler på allerede okkuperte steder ikke fører til en adsorpsjonshendelse. Denne antagelsen tilsvarer postulatet om monolagsadsorpsjon, og som vi sa tidligere, er den faktisk oppfylt i tilfellet med kjemisk adsorpsjon, men situasjonen er mer komplisert for fysisk adsorpsjon.
En annen antakelse gjort i utledningen av Henry isoterm-ligningen er at overflaten er homogen, dvs. ekvivalens av alle dens seksjoner, vil vi beholde uendret. Og til slutt, den tredje antakelsen i den nye modellen under vurdering er fraværet av interaksjon mellom adsorberte molekyler, dvs. vi vil anta at oppholdstiden til et molekyl på overflaten ikke er avhengig av hvor det treffer – i umiddelbar nærhet av et annet molekyl eller i stor avstand fra det. Alle disse antakelsene ble gjort av Langmuir i utledningen av adsorpsjonsisotermen, som han gjorde i 1918.
Langmuirs adsorpsjonsisotermligning kan utledes på forskjellige måter. Langmuir selv utledet det ved å vurdere avhengigheten av adsorpsjons- og desorpsjonshastighetene på graden av overflatedekning og anta at ved likevekt blir begge hastighetene de samme.
Den termodynamiske utledningen av denne ligningen ble gitt av Volmer, og den statistiske utledningen av Fowler.
I denne formen er Langmuir-ligningen viden kjent. Den inneholder to konstanter: en m kort kalt monolagkapasitet (maksimal adsorpsjon), og K er en konstant avhengig av adsorpsjonsenergi og temperatur.
adsorpsjon isoterm. Freundlich ligning.
Adsorpsjonsverdi (absolutt OG eller overskudd G) i hvert tilfelle bestemmes av temperaturen T og trykk R(med et gassformig adsorptivmiddel) eller temperatur T og konsentrasjon Med(når adsorbert fra løsninger). Som regel, i teorien om adsorpsjon, når man vurderer adsorpsjonslikevekten, holdes en av disse parameterne konstant. Så, en ligning av formen A \u003d f (p) T eller G \u003d f (c) T, som relaterer mengden av adsorpsjon med trykk eller konsentrasjon ved en konstant temperatur, kalles adsorpsjonsisotermen. Adsorpsjon (hvis det ikke uttrykkes som et overskudd, men som et totalt innhold) øker alltid med økende likevektstrykk eller konsentrasjon. Siden adsorpsjon er en eksoterm prosess, når temperaturen stiger, verdien adsorpsjon avtar. På fig. 26.9 viser hovedtypene av adsorpsjonslikevektskurver. Adsorpsjonsisotermer ved tre temperaturer (T 1 > T 2 > T 3) tilsvarer fig. 26,9a.
Figur 26.9. Kurver for adsorpsjonslikevekt: isotermer (a), isobarer (b) og isosterer (c) av adsorpsjon
Ligning som relaterer adsorpsjonsverdi til temperatur ved konstant likevektstrykk A \u003d f (T) s eller konstant likevektskonsentrasjon G \u003d f (T) s, kalles henholdsvis adsorpsjonsisobarer eller isopycnes (fig. 26.9-b); her p 1 > p 2 > p 3. Skriv ligning R= f (T) A, adsorpsjonsisosteret (fig. 26.9-c), relaterer likevektstrykket til temperaturen ved en konstant adsorbert mengde; i dette tilfellet A 1 > A 2 > A 3.
Oppgaven til enhver adsorpsjonsteori er å sammenstille dens matematiske beskrivelse på grunnlag av en bestemt modell av adsorpsjonsprosessen. Ideelt sett bør ligningen beskrive avhengigheten av likevektsverdien for adsorpsjon av konsentrasjonen av adsorbatet i bulkfasen ved forskjellige temperaturer, samt forutsi endringen i adsorpsjonsvarmen avhengig av fyllingen av adsorbenten. Oftest er adsorpsjonsisotermligningen funnet i dette tilfellet. Formen på adsorpsjonsisotermen på faste stoffer avhenger av mange parametere: egenskapene til adsorbenten og adsorbatet, interaksjonen av adsorbentadsorbatet, interaksjonen av adsorbatmolekyler med hverandre i gassfasen og i adsorbert tilstand. I området med lave trykk (eller konsentrasjoner) og de tilsvarende små overflatedekningene er interaksjonen mellom adsorbatmolekylene ubetydelig og avhengigheten A = f(p) T redusert til sin enkleste form, kalt Henrys lov:
A \u003d kp eller A \u003d k "c(26.20)
hvor k og til"- adsorpsjonskoeffisient (eller Henry-koeffisient), med er adsorbentkonsentrasjonen i bulkfasen, R er damptrykket til adsorbatet. Henry koeffisient k er et mål på intensiteten av adsorpsjon. Det kan vises at enhver teoretisk isoterm bør i grensen (for små fyllinger) gå over i Henry-ligningen.
I området medium konsentrasjoner, er konsentrasjonsavhengigheten av adsorpsjonen av oppløste stoffer godt beskrevet av empirien Freundlich ligning:
![]() (26.21)
(26.21)
hvor X- mengde adsorbert stoff, m- adsorberende masse, βi P - konstanter karakteristiske for hvert adsorpsjonssystem, med 0< 1/n< 1 . I følge Freindlich, n er ikke avhengig av polstring, selv om denne uttalelsen ikke er helt nøyaktig. Denne empiriske ligningen brukes ofte for omtrentlige beregninger av adsorpsjon. Oftest brukes det i logaritmisk form:
som tillater å konstruere en lineær avhengighet ln OG - ln c og grafisk bestemme begge konstante parametere β og n.
Overflatefenomener og adsorpsjon. Typer adsorpsjonsinteraksjoner. Gassadsorpsjonsisotermer. Henry og Langmuir-ligningen. Polymolekylær adsorpsjon, BET-teori.
OVERFLATEFENOMEN OG ADSORPJON
overflateenergi. Adsorpsjon
Til nå har egenskapene til heterogene systemer blitt beskrevet ved hjelp av parametere og tilstandsfunksjoner som karakteriserer hver av fasene som helhet. Imidlertid avviker egenskapene til faseområdet ved siden av overflaten fra egenskapene til fasen i volumet: faktisk danner partiklene på overflaten av hver fase en spesiell overflatefase, hvis egenskaper skiller seg betydelig fra egenskapene av de indre områdene i fasen. Partiklene som ligger på overflaten er i et annet miljø sammenlignet med partiklene som befinner seg i fasens volum, dvs. interagerer både med homogene partikler og med partikler av en annen type. Konsekvensen av dette er at den gjennomsnittlige energien g s til en partikkel som befinner seg på fasegrensesnittet skiller seg fra den gjennomsnittlige energien til den samme partikkelen i volumet til fasen g v (dessuten kan energien til en partikkel på overflaten være enten større eller mindre enn energien til en partikkel i volumet). Derfor er den viktigste egenskapen til overflatefasen overflateenergi G s er forskjellen mellom den gjennomsnittlige energien til en partikkel lokalisert på overflaten og en partikkel lokalisert i volumet av fasen, multiplisert med antall partikler på overflaten N:
![]() (26.1)
(26.1)
Det er åpenbart at den totale verdien av overflateenergien til en fase vil bli bestemt av verdien av dens overflate S. Derfor, for å karakterisere grensesnittet som skiller en gitt fase fra en annen, introduseres konseptet overflatespenningσ er forholdet mellom overflateenergien og arealet av grensesnittet; størrelsen på overflatespenningen avhenger bare av naturen til begge fasene. I likhet med overflateenergien til en fase, kan overflatespenningen være enten positiv eller negativ. Overflatespenningen er positiv hvis partiklene på overflaten samhandler med partikler av samme fase sterkere enn med partikler av en annen fase (og derfor g s > g v). I henhold til prinsippet om minimum fri energi, vil enhver fase ha en tendens til spontant å redusere overflateenergien; derfor, i tilfelle av positiv overflatespenning (σ > 0), har fasen en tendens til å redusere overflaten. Hvis σ< 0, поверхностная энергия фазы будет уменьшаться при увеличении площади поверхности.
Påvirkningen av overflatelaget til en fase på dens generelle egenskaper bestemmes av andelen partikler lokalisert på overflaten av det totale antallet partikler som utgjør denne fasen, dvs. verdien av den spesifikke overflaten til fasen S/V (overflate per volumenhet). Den frie energien til fasen G kan representeres som summen av overflaten G s og volumet G v energier proporsjonal med overflatearealet og volumet til fasen, henholdsvis:
Ved å dele dette uttrykket med volumet av fasen får vi:
Det følger av ligning (IV.4) at med samme mengde fase (dvs. konstant volum), øker overflateenergiens bidrag til fasens totale energi med en økning i det spesifikke overflatearealet, eller i andre ord, grad av spredning(fragmenterings)fase. I tilfellet når spredningsgraden av fasen er liten (den spesifikke overflaten er ubetydelig), neglisjeres vanligvis bidraget fra overflateenergien til den totale energien til fasen. Overflatelagets bidrag til egenskapene til fasen og systemet som helhet tas i betraktning når man studerer spre systemer- heterogene systemer, hvor en av fasene er kontinuerlig ( dispersjonsmedium), og den andre er fragmentert ( spredt fase).
Ved grensen til en kondensert (dvs. fast eller flytende) fase med en gass er overflatespenningen alltid positiv, siden partiklene i den kondenserte fasen interagerer sterkere med hverandre enn med gassmolekyler. I henhold til prinsippet om minimum fri energi, vil den kondenserte fasen ha en tendens til spontant å redusere overflateenergien. Dette kan være resultatet av enten en reduksjon i overflatearealet til fasen (det er derfor en dråpe væske i vektløshet har form av en kule), eller en reduksjon i overflatespenning når nye partikler vises på fasegrensesnittet - gassmolekyler eller et oppløst stoff. Prosessen med spontan endring i konsentrasjonen av et stoff i grensesnittet mellom to faser kalles adsorpsjon. Adsorbent et stoff kalles, på overflaten som det er en endring i konsentrasjonen av et annet stoff - adsorbat.
Adsorpsjon ved løsning-damp-grensesnittet
I flytende løsninger er overflatespenningen σ en funksjon av konsentrasjonen av oppløst stoff. På fig. 4.1 viser tre mulige avhengigheter av overflatespenning av konsentrasjonen av løsningen (de såkalte overflatespenningsisotermene). Stoffer hvis tilsetning til et løsemiddel reduserer overflatespenningen kalles overflateaktiv(overflateaktive stoffer), stoffer hvis tilsetning øker eller ikke endrer overflatespenningen - overflate-inaktiv(PIAV).

Ris. 26.1 Overflateisotermer 26.2 Adsorpsjonsisoterm
spenning av overflateaktive løsninger (1, 2) og PIAV. Overflateaktivt middel ved løsning-damp-grensesnittet
PIAV (3)
En reduksjon i overflatespenning og følgelig overflateenergi oppstår som et resultat av surfaktantadsorpsjon på væske-damp-grensesnittet, dvs. det faktum at konsentrasjonen av overflateaktivt middel i overflatelaget til løsningen er større enn i løsningens dybde.
Det kvantitative målet for adsorpsjon ved løsning-damp-grensesnittet er overflateoverskudd G (gamma), lik antall mol oppløst stoff i overflatelaget. Det kvantitative forholdet mellom adsorpsjonen (overflateoverskuddet) av et oppløst stoff og endringen i overflatespenningen til oppløsningen med økende oppløsningskonsentrasjon bestemmer Gibbs adsorpsjonsisoterm:
Plottet for adsorpsjonsisotermen for overflateaktivt middel er vist i fig. 26.2. Det følger av ligning (26.5) at retningen av prosessen - konsentrasjonen av et stoff i overflatelaget eller omvendt dets tilstedeværelse i hoveddelen av væskefasen - bestemmes av tegnet til derivatet dσ / dС. Den negative verdien av dette derivatet tilsvarer akkumuleringen av stoffet i overflatelaget (G > 0), den positive verdien tilsvarer en lavere konsentrasjon av stoffet i overflatelaget sammenlignet med dets konsentrasjon i hoveddelen av løsningen.
Verdien g = –dσ/dС kalles også overflateaktiviteten til det oppløste stoffet. Overflateaktiviteten til overflateaktive stoffer ved en viss konsentrasjon av C 1 bestemmes grafisk ved å tegne en tangent til overflatespenningsisotermen i punktet C = C 1; i dette tilfellet er overflateaktiviteten numerisk lik tangenten til helningen til tangenten til konsentrasjonsaksen:
Det er lett å se at med økende konsentrasjon avtar overflateaktiviteten til overflateaktive stoffer. Derfor er overflateaktiviteten til et stoff vanligvis bestemt ved en uendelig liten konsentrasjon av løsningen; i dette tilfellet avhenger dens verdi, betegnet g o, bare av naturen til det overflateaktive midlet og løsningsmidlet. Ved å undersøke overflatespenningen til vandige løsninger av organiske stoffer, etablerte Traube og Duclos følgende tommelfingerregel for den homologe serien av overflateaktive stoffer:
I enhver homolog serie ved lave konsentrasjoner øker forlengelsen av karbonkjeden med én CH2-gruppe overflateaktiviteten med en faktor på 3–3,5.
For vandige løsninger av fettsyrer er overflatespenningens avhengighet av konsentrasjon beskrevet av empirien. Shishkovsky-ligningen:
Her er b og K empiriske konstanter, og verdien av b er den samme for hele den homologiske serien, og verdien av K øker for hvert påfølgende medlem av serien med 3–3,5 ganger.

Ris. 26.3 Begrens orienteringen av overflateaktive molekyler i overflatelaget
Molekyler av de fleste overflateaktive stoffer har en amfifil struktur, dvs. inneholder både en polar gruppe og et ikke-polart hydrokarbonradikal. Plasseringen av slike molekyler i overflatelaget er energetisk mest gunstig under forutsetning av at molekylene er orientert av den polare gruppen til den polare fasen (polar væske), og den upolare gruppen til den ikke-polare fasen (gass eller ikke-polar væske). Ved lav konsentrasjon av løsningen forstyrrer termisk bevegelse orienteringen til overflateaktive molekyler; med en økning i konsentrasjonen mettes adsorpsjonslaget og det dannes et lag med "vertikalt" orienterte overflateaktive molekyler på grensesnittet (fig. 26.3). Dannelsen av et slikt monomolekylært lag tilsvarer minimumsverdien av overflatespenningen til den overflateaktive løsningen og maksimalverdien av adsorpsjon G (fig. 26.1-26.2); med en ytterligere økning i konsentrasjonen av overflateaktivt middel i løsningen, endres ikke overflatespenningen og adsorpsjonen.
Adsorpsjon ved fast-gass-grensesnittet
Når gasser adsorberes på faste stoffer, er beskrivelsen av interaksjonen mellom adsorbat- og adsorbentmolekyler et svært komplekst problem, siden arten av deres interaksjon, som bestemmer arten av adsorpsjon, kan være forskjellig. Derfor blir problemet vanligvis forenklet ved å vurdere to ekstreme tilfeller, når adsorpsjon er forårsaket av fysiske eller kjemiske krefter – henholdsvis fysisk og kjemisk adsorpsjon.
fysisk adsorpsjon oppstår på grunn av van der Waals-interaksjoner. Det er preget av reversibilitet og en reduksjon i adsorpsjon med økende temperatur, dvs. eksotermisitet, og varmeeffekten av fysisk adsorpsjon er vanligvis nær varmen fra flytendegjøring av adsorbatet (10 – 80 kJ/mol). Slik er for eksempel adsorpsjon av inerte gasser på kull.
Kjemisk adsorpsjon(kjemisorpsjon) utføres ved kjemisk interaksjon av adsorbent- og adsorbatmolekyler. Kjemisorpsjon er vanligvis irreversibel; kjemisk adsorpsjon, i motsetning til fysisk adsorpsjon, er lokalisert; adsorbatmolekyler kan ikke bevege seg over overflaten av adsorbenten. Siden kjemisorpsjon er en kjemisk prosess som krever en aktiveringsenergi på ca. 40-120 kJ/mol, bidrar en temperaturøkning til at den oppstår. Et eksempel på kjemisk adsorpsjon er adsorpsjon av oksygen på wolfram eller sølv ved høye temperaturer.
Det bør understrekes at fenomenene fysisk og kjemisk adsorpsjon er tydelig skilt i svært sjeldne tilfeller. Mellomalternativer utføres vanligvis når hoveddelen av det adsorberte stoffet binder seg relativt svakt og bare en liten del er fast bundet. For eksempel blir oksygen på metaller eller hydrogen på nikkel ved lave temperaturer adsorbert i henhold til lovene for fysisk adsorpsjon, men når temperaturen stiger, begynner kjemisk adsorpsjon å skje. Når temperaturen stiger, begynner økningen i kjemisk adsorpsjon fra en viss temperatur å overlappe fallet i fysisk adsorpsjon, slik at temperaturavhengigheten av adsorpsjon i dette tilfellet har et klart definert minimum (fig. 26.4).

Ris. 26.4 Temperaturens avhengighet av volumet av hydrogen adsorbert av nikkel
Ved konstant temperatur avhenger mengden av adsorbert stoff kun av likevektstrykket eller konsentrasjonen av adsorbatet; ligningen som relaterer disse mengdene kalles adsorpsjonsisotermen.
Teorier om adsorpsjon
Det er ingen enhetlig teori som godt kan beskrive alle typer adsorpsjon på forskjellige fasegrensesnitt; La oss derfor vurdere noen av de vanligste adsorpsjonsteoriene som beskriver individuelle typer adsorpsjon på grensesnittet fast-gass eller fast-løsning.
Langmuirs teori om monomolekylær adsorpsjon
Teorien om monomolekylær adsorpsjon, som ble utviklet av den amerikanske kjemikeren I. Langmuir, er basert på følgende bestemmelser.
1) Adsorpsjon er lokalisert og er forårsaket av krefter nær kjemiske.
2) Adsorpsjon skjer ikke på hele overflaten av adsorbenten, men på aktive sentre, som er fremspring eller fordypninger på overflaten av adsorbenten, preget av tilstedeværelsen av den såkalte. gratis valenser. Aktive sentre anses som uavhengige (dvs. ett aktivt senter påvirker ikke adsorpsjonskapasiteten til andre), og identiske.
3) Hvert aktivt senter er kun i stand til å samhandle med ett adsorbatmolekyl; som et resultat kan bare ett lag med adsorberte molekyler dannes på overflaten.
4) Adsorpsjonsprosessen er reversibel og balansert– det adsorberte molekylet beholdes av det aktive senteret i noen tid, hvoretter det desorberes; dermed, etter en tid, etableres en dynamisk likevekt mellom adsorpsjons- og desorpsjonsprosessene.

Ris. 26.5 Monomolekylær adsorpsjonsisoterm
I likevektstilstanden er adsorpsjonshastigheten lik desorpsjonshastigheten. Desorpsjonshastigheten er direkte proporsjonal med andelen av okkuperte aktive sentre (x), og adsorpsjonshastigheten er direkte proporsjonal med produktet av adsorbatkonsentrasjonen og fraksjonen av frie aktive sentre (1 – x):
![]() (26.9)
(26.9)
Herfra finner vi x:
Ved å dele telleren og nevneren til høyre side av ligningen (26.10) med k A , får vi:
 (26.11)
(26.11)
Maksimal mulig verdi av adsorpsjon To oppnås under forutsetning av at alle aktive sentre er okkupert av adsorbatmolekyler, dvs. x = 1. Derfor følger det at x = r / r o. Setter vi dette inn i ligningen (26.11), får vi:
Ligning (26.13) er monomolekylær adsorpsjonsisoterm, som relaterer verdien av adsorpsjon G til konsentrasjonen av adsorbat C. Her er b en viss konstant verdi for et gitt adsorbent-adsorbat-par (forholdet mellom desorpsjons- og adsorpsjonshastighetskonstanter), numerisk lik adsorbatkonsentrasjonen, hvor halvparten av de aktive sentrene er okkupert. Rute Langmuir adsorpsjonsisotermer vist i fig. 26.5. Konstanten b kan bestemmes grafisk ved å tegne en tangent til adsorpsjonsisotermen i punktet C = 0.
Når man beskriver prosessen med adsorpsjon av gasser i ligning (26.13), kan konsentrasjonen erstattes med en proporsjonal verdi av gassens partialtrykk:
Langmuirs teori om monomolekylær adsorpsjon er anvendelig for å beskrive noen prosesser for adsorpsjon av gasser og oppløste stoffer ved lave trykk (konsentrasjoner) av adsorbatet.
Polanyis teori om polymolekylær adsorpsjon
I praksis er det ofte (spesielt ved adsorpsjon av damper) såkalte. S-formede adsorpsjonsisotermer (fig. 4.6), hvis form indikerer mulig, med utgangspunkt i en viss trykkverdi, interaksjonen av adsorberte molekyler med adsorbatet.

Ris. 26.6 Polymolekylær adsorpsjonsisoterm
For å beskrive slike adsorpsjonsisotermer foreslo M. Polyani teori om polymolekylær adsorpsjon basert på følgende hovedprinsipper:
1. Adsorpsjon forårsaket rent fysiske krefter.
2. Adsorberende overflate homogen, dvs. det er ingen aktive sentre på den; adsorpsjonskrefter danner et kontinuerlig kraftfelt nær overflaten av adsorbenten.
3. Adsorpsjonskrefter virker i en avstand større enn størrelsen på adsorbatmolekylet. Med andre ord, på overflaten av adsorbenten er det noe adsorpsjonsvolum, som er fylt med adsorbatmolekyler under adsorpsjon.
4. Tiltrekningen av et adsorbatmolekyl av adsorbentoverflaten er ikke avhengig av tilstedeværelsen av andre molekyler i adsorpsjonsvolumet, som et resultat av at det er mulig polymolekylær adsorpsjon.
5. Adsorpsjonskrefter ikke avhengig av temperaturen og følgelig, med en endring i temperatur, endres ikke adsorpsjonsvolumet.
Freundlich ligning
De teoretiske konseptene utviklet av Langmuir og Polanyi idealiserer og forenkler i stor grad det sanne bildet av adsorpsjon. Faktisk er overflaten av adsorbenten inhomogen, det er en interaksjon mellom de adsorberte partiklene, aktive sentre er ikke helt uavhengige av hverandre, etc. Alt dette kompliserer formen til isotermligningen. G. Freindlich viste at ved en konstant temperatur er antall mol adsorbert gass eller oppløst stoff per masseenhet av adsorbenten (den såkalte spesifikke adsorpsjonen x/m) proporsjonal med likevektstrykket (for gass) eller likevektskonsentrasjonen ( for stoffer adsorbert fra løsning) av adsorbenten hevet til en viss styrke, som alltid er mindre enn én:
Adsorpsjon ved grensesnittet med fast løsning
Molekylær adsorpsjon fra løsninger
Adsorpsjonsisotermer av oppløste stoffer fra en løsning ligner i utseende på adsorpsjonsisotermer for gasser; for fortynnede løsninger er disse isotermene godt beskrevet av Freundlich- eller Langmuir-ligningene, hvis likevektskonsentrasjonen til det oppløste stoffet i løsningen erstattes med dem. Imidlertid er adsorpsjon fra løsninger et mye mer komplekst fenomen sammenlignet med gass, siden adsorpsjonen av et løsningsmiddel ofte skjer samtidig med adsorpsjonen av et løst stoff.

Ris. 26.8 Orientering av overflateaktive molekyler på den adsorberende overflaten
Avhengigheten av adsorpsjon av strukturen til adsorbatmolekyler er svært kompleks, og det er ganske vanskelig å utlede noen regulariteter. Molekyler av mange organiske stoffer består av polare (hydrofile) og ikke-polare (hydrofobe) grupper, dvs. er overflateaktive stoffer. Når de adsorberes på en fast adsorbent, er overflateaktive molekyler orientert på overflaten på en slik måte at den polare delen av molekylet vender mot den polare fasen, og den ikke-polare delen vender mot den ikke-polare fasen. Så, under adsorpsjonen av alifatiske karboksylsyrer fra vandige løsninger på en ikke-polar adsorbent - aktivert karbon - blir molekylene orientert av hydrokarbonradikaler mot adsorbenten; når adsorbert fra benzen (ikke-polart løsningsmiddel) på en polar adsorbent - silikagel - vil orienteringen til syremolekylene bli reversert (fig. 4.8).
Adsorpsjon fra elektrolyttløsninger
Adsorpsjon fra vandige oppløsninger av elektrolytter skjer som regel på en slik måte at ioner av samme type adsorberes fra oppløsningen på den faste adsorbenten. Foretrukket adsorpsjon fra en løsning eller et anion eller et kation bestemmes av arten av adsorbenten og ionene. Mekanismen for adsorpsjon av ioner fra elektrolyttløsninger kan være forskjellig; allokere utveksling og spesifikk adsorpsjon av ioner.
Utvekslingsadsorpsjon er en ioneutvekslingsprosess mellom en løsning og en fast fase, der den faste fasen absorberer ioner av et bestemt tegn (kationer eller anioner) fra løsningen og i stedet frigjør et ekvivalent antall andre ioner med samme fortegn inn i løsningen. Utvekslingsadsorpsjon er alltid spesifikk, dvs. for en gitt adsorbent er bare visse ioner i stand til å bytte; utvekslingsadsorpsjon er vanligvis irreversibel.
På spesifikk adsorpsjon adsorpsjon på overflaten av den faste fasen av ioner av noe slag er ikke ledsaget av frigjøring i løsningen av et ekvivalent antall andre ioner av samme fortegn; den faste fasen får en elektrisk ladning. Dette fører til det faktum at nær overflaten, under påvirkning av elektrostatiske tiltrekningskrefter, grupperes et ekvivalent antall ioner med motsatte ladninger, dvs. det dannes et elektrisk dobbeltlag. Samspillet mellom ladninger som konsentrerer seg om overflaten fører til en reduksjon i overflateenergien til systemet. For spesifikk elektrolyttadsorpsjon formulerte Peskov og Fayans følgende empiriske regel ( Peskov-Faience regel):
På overflaten av et krystallinsk fast stoff adsorberes et ion spesifikt fra en elektrolyttløsning, som er i stand til å fullføre krystallgitteret eller kan danne en dårlig løselig forbindelse med et av ionene som utgjør krystallen.
Henrys lov kan formuleres som følger: når systemet fortynnes (trykket synker), tenderer fordelingskoeffisienten til en konstant verdi lik Henrys fordelingskonstanten. Med hensyn til adsorpsjonsverdien A kan denne loven skrives som følger:
E  Disse ligningene er adsorpsjonsisotermer av et stoff i lave konsentrasjoner. I samsvar med dem kan Henrys lov formuleres som følger: mengden adsorpsjon ved lave gasstrykk (stoffkonsentrasjoner i løsning) er direkte proporsjonal med trykk (konsentrasjon).
Disse ligningene er adsorpsjonsisotermer av et stoff i lave konsentrasjoner. I samsvar med dem kan Henrys lov formuleres som følger: mengden adsorpsjon ved lave gasstrykk (stoffkonsentrasjoner i løsning) er direkte proporsjonal med trykk (konsentrasjon).
Avvik fra Henrys lov, uttrykt ved endringer i aktivitetskoeffisientene i fasene, tillater vanligvis ikke at man kan beskrive og forutsi forløpet av isotermer med økende konsentrasjon.
(trykk) av adsorbatet. For å oppnå en teoretisk adsorpsjonsisoterm som beskriver et bredere spekter av konsentrasjoner, er det nødvendig å bruke konsepter for adsorpsjonsmekanismen og spesifikke modeller.
En stor brøkdel av avvik av adsorbataktivitetskoeffisienten i overflatelaget fra enhet kan tas i betraktning ved å bruke ideen om adsorpsjon som en kvasikjemisk reaksjon mellom adsorbatet og adsorpsjonssentrene til adsorbentoverflaten. Dette er hovedideen til Langmuirs adsorpsjonsteori. Denne bestemmelsen er tydeliggjort av følgende forutsetninger:
1) adsorpsjon er lokalisert (molekyler beveger seg ikke over overflaten) på separate adsorpsjonssentre, som hver samhandler med bare ett adsorbatmolekyl; som et resultat dannes et monomolekylært lag;
2) adsorpsjonssentre er energisk ekvivalente - overflaten av adsorbenten er ekvipotensial;
3) adsorberte molekyler interagerer ikke med hverandre.
Lyofile spredningssystemer. Klassifisering og generelle egenskaper ved pav. Termodynamikk og micelliseringsmekanisme. Strukturen til overflateaktive miceller i vandige og hydrokarbonmedier. Solubilisering.
Alle dispergerte systemer, avhengig av mekanismen for deres dannelse, i henhold til klassifiseringen av P. A. Rebinder, er delt inn i lyofile, som oppnås ved spontan dispersjon av en av fasene (spontan dannelse av et heterogent fritt dispergert system), og lyofobe. , som følge av spredning og kondensasjon med overmetning (tvungen dannelse av et heterogent frittgående system).
Hvis overflatespenningen ved grensesnittet avtar med økende konsentrasjon av et stoff, kalles et slikt stoff overflateaktivt. For slike stoffer er overflateaktiviteten
Tilstedeværelsen av hydrofile og oleofile deler i overflateaktive molekyler er et karakteristisk kjennetegn ved deres struktur. I henhold til evnen til å dissosiere i vandige løsninger, er overflateaktive stoffer delt inn i ioniske og ikke-ioniske. I sin tur er ioniske overflateaktive stoffer delt inn i anioniske, kationiske og amfolytiske (amfotere).
1) Anioniske overflateaktive stoffer dissosieres i vann for å danne et overflateaktivt anion.
2) Kationiske overflateaktive stoffer dissosieres i vann for å danne et overflateaktivt kation.
3) Amfolytiske overflateaktive stoffer inneholder to funksjonelle grupper, hvorav den ene er sur og den andre basisk, som karboksyl- og amingrupper. Avhengig av pH i mediet, viser amfolytiske overflateaktive stoffer anioniske eller kationiske egenskaper.
Alle overflateaktive stoffer med hensyn til deres oppførsel i vann er delt inn i virkelig løselige og kolloidale.
Virkelig løselige overflateaktive stoffer i løsning er i en molekylært dispergert tilstand opp til konsentrasjoner som tilsvarer deres mettede løsninger og separasjon av systemet i to kontinuerlige faser.
Det viktigste kjennetegnet ved kolloidale overflateaktive stoffer er evnen til å danne termodynamisk stabile (lyofile) heterogene disperse systemer (assosiative eller micellære, kolloider). Hovedegenskapene til kolloidale overflateaktive stoffer, som bestemmer deres verdifulle kvaliteter og brede anvendelse, inkluderer høy overflateaktivitet; evnen til spontan micelledannelse - dannelsen av lyofile kolloidale løsninger ved en overflateaktivt konsentrasjon over en viss spesifikk verdi, kalt den kritiske micellekonsentrasjonen (KKM); evnen til å solubilisere - en kraftig økning i løseligheten av stoffer i løsninger av kolloidale overflateaktive stoffer på grunn av deres "introduksjon" i micellene; høy evne til å stabilisere ulike spredningssystemer.
Ved konsentrasjoner over KKM samles overflateaktive molekyler til miceller (assosierte) og løsningen transformeres til et micellært (assosiativt) kolloidalt system.
En overflateaktiv micelle forstås som en assosiasjon av amfifile molekyler, hvis lyofile grupper vender mot det tilsvarende løsningsmidlet, og de lyofobe gruppene er koblet til hverandre, og danner kjernen til micellen. Antall molekyler som utgjør en micelle kalles assosiasjonsnummeret, og den totale summen av molekylvektene til molekylene i micellen, eller produktet av massen til micellen og Avogadro-tallet, kalles micellemassen. En viss orientering av amfifile overflateaktive molekyler i en micelle gir en minimum grensesnittspenning ved micelle-miljøgrensen.
P  Ved konsentrasjoner av overflateaktive stoffer i en vandig løsning som er noe høyere enn KKM, dannes det ifølge Hartleys konsepter sfæriske miceller (Hartley-miceller). Den indre delen av Gartley-miceller består av sammenflettede hydrokarbonradikaler, de polare gruppene av overflateaktive molekyler omdannes til den vandige fasen. Diameteren til slike miceller er lik to ganger lengden på overflateaktive molekyler. Antall molekyler i en micelle vokser raskt innenfor et smalt konsentrasjonsområde, og med ytterligere økning i konsentrasjonen endres det praktisk talt ikke, men antallet miceller øker. Sfæriske miceller kan inneholde fra 20 til 100 molekyler eller mer.
Ved konsentrasjoner av overflateaktive stoffer i en vandig løsning som er noe høyere enn KKM, dannes det ifølge Hartleys konsepter sfæriske miceller (Hartley-miceller). Den indre delen av Gartley-miceller består av sammenflettede hydrokarbonradikaler, de polare gruppene av overflateaktive molekyler omdannes til den vandige fasen. Diameteren til slike miceller er lik to ganger lengden på overflateaktive molekyler. Antall molekyler i en micelle vokser raskt innenfor et smalt konsentrasjonsområde, og med ytterligere økning i konsentrasjonen endres det praktisk talt ikke, men antallet miceller øker. Sfæriske miceller kan inneholde fra 20 til 100 molekyler eller mer.
Når konsentrasjonen av overflateaktivt middel øker, går det micellære systemet gjennom en rekke likevektstilstander som er forskjellige i assosiasjonsantall, størrelser og former for miceller. Når en viss konsentrasjon er nådd, begynner sfæriske miceller å samhandle med hverandre, noe som bidrar til deres deformasjon. Miceller har en tendens til å ha en sylindrisk, skiveformet, stavformet, lamellformet form.
Micellisering i ikke-vandige medier er som regel resultatet av virkningen av tiltrekningskrefter mellom de polare gruppene av overflateaktive stoffer og samspillet mellom hydrokarbonradikaler og løsemiddelmolekyler. De inverterte micellene som dannes inneholder ikke-hydratiserte eller hydratiserte polare grupper inni, omgitt av et lag med hydrokarbonradikaler. Assosiasjonstallet (fra 3 til 40) er mye mindre enn for vandige løsninger av overflateaktive stoffer. Som regel vokser det med en økning i hydrokarbonradikalet opp til en viss grense.
Fenomenet med oppløsning av stoffer i overflateaktive miceller kalles solubilisering. Måten solubilisatmolekyler er inkludert i miceller i vandige løsninger avhenger av stoffets natur. Ikke-polare hydrokarboner, som trenger inn i miceller, er lokalisert i hydrokarbonkjernene til miceller. Polare organiske stoffer (alkoholer, aminer, syrer) inkorporeres i en micelle mellom overflateaktive molekyler slik at deres polare grupper vender mot vann, og de lipofile delene av molekylene er orientert parallelt med de overflateaktive hydrokarbonradikalene. En tredje måte å inkorporere solubilisatet i miceller er også mulig, som er spesielt karakteristisk for ikke-ioniske overflateaktive stoffer. Molekyler av et solubilisat, slik som fenol, trenger ikke inn i miceller, men er fiksert på overflaten, plassert mellom tilfeldig bøyde polyoksyetylenkjeder.
Solubilisering er en spontan og reversibel prosess; en gitt overflateaktivt konsentrasjon og temperatur tilsvarer en veldefinert metning av løsningen med solubilisat. Som et resultat av solubilisering oppnås stabile disperse systemer som ligner spontant dannede ultramikroheterogene emulsjoner.
Bestem overflaten og den totale (interne) energien til 4 g vanntåke, som har partikler med en dispersjon på 5 10 7 m -1 , t= 20ºC, o = 72 mJ/m 2 ; dσ/ dT= - 0,16 mJ/(m 2 ·TIL); ρ = 1000 kg/m 3 .

Eksamensbillett nummer 10
Teori om polymolekylær BET-adsorpsjon: startposisjoner, utledning av isotermligningen og dens analyse. Lineær form av BET-ligningen. Bestemmelse av den spesifikke overflaten til adsorbenter, katalysatorer og andre porøse legemer.
Langmuir-ligningen kan bare brukes under forutsetning av at adsorpsjonen av et stoff er ledsaget av dannelsen av et monomolekylært lag.
I de fleste tilfeller kompenserer ikke et monomolekylært adsorpsjonslag fullt ut for overflødig overflateenergi, og påvirkningen av overflatekrefter kan strekke seg til det andre, tredje og påfølgende adsorpsjonslaget, noe som resulterer i polymolekylær adsorpsjon.
Med  Den moderne formen for den polymolekylære adsorpsjonsligningen - den grunnleggende ligningen til den generaliserte Langmuir-teorien - ble foreslått av Brunauer, Emmett og Teller.
Den moderne formen for den polymolekylære adsorpsjonsligningen - den grunnleggende ligningen til den generaliserte Langmuir-teorien - ble foreslått av Brunauer, Emmett og Teller.
I denne teorien er en tilleggsantagelse til de som ble brukt som grunnlag for utledningen av Langmuir-isotermligningen ideen om dannelsen av "påfølgende komplekser" av adsorpsjonssentre med en, to, tre, etc. adsorbatmolekyler på overflaten av adsorbenten. Da kan adsorpsjonsprosessen representeres som påfølgende kvasikjemiske reaksjoner:
Likevektskonstantene til disse reaksjonene er henholdsvis lik
Betegn:
Det totale antallet aktive steder på adsorbenten, eller kapasiteten til monolaget, vil være lik

Etter en rekke beregninger ved hjelp av serieteorien får vi til slutt:

D  Denne relasjonen er den grunnleggende ligningen til den generaliserte Langmuir-teorien og kalles BET polymolekylær adsorpsjonsligning.
Denne relasjonen er den grunnleggende ligningen til den generaliserte Langmuir-teorien og kalles BET polymolekylær adsorpsjonsligning.
Når du behandler eksperimentelle resultater, brukes BET-ligningen vanligvis i en lineær form:
Den lar deg grafisk bestemme både konstante parametere A ∞ og С:
Den eksperimentelle bestemmelsen av A ∞ gjør det mulig å beregne det spesifikke overflatearealet til adsorbenten (overflateareal per masseenhet av adsorbenten): ![]() .
.
Adsorpsjon- konsentrasjon av et stoff fra volumet av faser på grensesnittet mellom dem. Adsorpsjon kan sees som absorpsjon substans (adsorbat) overflate av adsorbenten.
Adsorbent Stoffet hvis overflate adsorpsjon finner sted.
Adsorbtiv - en gass eller oppløst stoff som er i stand til å bli adsorbert på overflaten av en adsorbent.
Adsorbat - adsorbert stoff på overflaten av adsorbenten. Ofte identifiseres begrepene "adsorbent" "adsorbat".
Skille fysisk adsorpsjon, skjer uten kjemisk endring av adsorbatet og kjemisk adsorpsjon(kjemisorpsjon), ledsaget av kjemisk interaksjon mellom adsorbenten og adsorbenten.
Adsorpsjon skjer ved fasegrensene: fast - væske, fast - gass, væske - gass, væske - væske.
Når et stoff adsorberes i form av molekyler, kalles det molekylær adsorpsjon, i form av ioner - ionisk adsorpsjon.
Adsorpsjon er reversibel, den omvendte prosessen kalles desorpsjon.
Hastighetene for adsorpsjon og desorpsjon er lik hverandre ved adsorpsjonslikevekt, som tilsvarer likevektskonsentrasjon adsorbat i løsning eller likevektstrykk i gassfasen.
Adsorpsjonsverdi(A) er karakterisert ved likevektsmengden av absorbert stoff (X) per masseenhet fast adsorbent (m): [mol/kg eller kg/kg]
Adsorpsjonsisoterm- grafisk fremstilling av adsorpsjonsverdiens avhengighet av likevektskonsentrasjonen eller likevektstrykket ved en gitt konstant temperatur.
Skille adsorpsjon monomolekylært, hvor adsorbatet dekker overflaten av adsorbenten med et lag ett molekyl tykt og polymolekylær, hvor adsorbatmolekylene kan lokaliseres på overflaten av adsorbenten i flere lag.
Monomolekylær adsorpsjonsisoterm har formen vist i fig. 12 ( Langmuir isoterm)
A Site I - svar liten likevektskonsentrasjoner (trykk), når en liten del av adsorbentoverflaten er okkupert av adsorbatmolekyler, og avhengigheten A - c (p) er lineær;
Seksjon II - medium konsentrasjoner (trykk) hvor en betydelig andel av adsorbentoverflaten er okkupert av adsorbatmolekyler;
c (p) Seksjon III - observert kl høy likevektskonsentrasjoner (trykk), når hele adsorbentoverflaten er okkupert av adsorbatmolekyler og nås grenseverdi for adsorpsjon (A).
Isoterm av monomolekylær adsorpsjonsbrønn er beskrevet av Langmuir-ligningen:

hvor c, A individuelle konstanter for hvert enkelt stoff under adsorpsjon på en spesifikk adsorbent;
s, r- likevektskonsentrasjon eller likevektstrykk.
Ved lave likevektskonsentrasjoner kan vi neglisjere verdien med eller R i nevneren. Deretter transformeres Langmuir-ligningen til ligningen til en rett linje som går gjennom origo:
A = A i c eller A \u003d A på s
På høye likevektskonsentrasjoner kan neglisjeres i nevneren i. Deretter transformeres Langmuir-ligningen til ligningen til en rett linje uavhengig av med eller R: A = A
For praktiske beregninger det er nødvendig å kjenne konstantene til Langmuir-ligningen A og i.Å transformere ligningen til en lineær form av en rett linje som ikke går gjennom origo for koordinater: , lar deg bygge en graf av avhengigheten 1/A - 1/c (fig. 13).
1/A Segmentet OB er lik 1/A. Koeffisient i kan finnes fra det faktum at i er lik konsentrasjonen hvor adsorpsjonsmengden er halvparten av grensen.

På grafen bestemmer interpolasjon segmentet OD som tilsvarer 2/A og lik 1/in. Da er s = 1/OD.
Langmuir-ligningen ble avledet fra teorien om monomolekylær adsorpsjon, som har følgende grunnleggende bestemmelser:
adsorpsjon av molekyler skjer bare på adsorpsjonssentre (topper av uregelmessigheter og trange porer);
hvert adsorpsjonssenter kan inneholde bare ett adsorbatmolekyl;
adsorpsjonsprosessen er reversibel; Adsorpsjonslikevekten er dynamisk. Adsorberte molekyler beholdes av adsorpsjonssentre bare i en viss tid, hvoretter disse molekylene desorberes og det samme antall nye molekyler adsorberes.
I tillegg til Langmuir-ligningen, brukes den i praksis ofte Freundlich ligning:
A \u003d KS 1 / n eller A \u003d KR 1 / n, hvor K og 1 / n er empiriske konstanter.
Ligningen er mer egnet til å beskrive adsorpsjon på porøs eller pulverisert adsorbenter i området gjennomsnittlige konsentrasjoner (trykk).
Freundlich-adsorpsjonsisotermen har ikke en horisontal rett linje og adsorpsjonen øker med økende konsentrasjon (trykk) (fig. 14).

Ris. fjorten
Til finne konstantene til Freundlich-ligningen den konverteres ved hjelp av logaritmen til ligningen til en rett linje som ikke går gjennom origo: lg A \u003d log K + 1 / n lg C.
I samsvar med dette har grafen for avhengigheten av lg A av lg C eller (P), bygget i henhold til eksperimentelle data, formen vist i fig. 15. Ved ekstrapolering til ordinataksen oppnås et segment OB lik lg K. Tangensen til helningsvinkelen til den rette linjen BN til abscisseaksen er 1/n ( tg =)
Polymolekylær adsorpsjon- observert under adsorpsjon på porøse eller pulveriserte adsorbenter (silikagel, aktivert kull, pulver og tabletter av medisinske stoffer). I dette tilfellet fortsetter adsorpsjonen til et tett monomolekylært lag er dannet, som vist i fig. 16.

Ris. 16.

Slik adsorpsjon tilsvarer en annen type isoterm (fig. 17), den såkalte " S - isoterm".
kapillær kondensasjon- Fenomenet damp flytendegjøring i porene eller kapillærene til en fast adsorbent, det observeres når lett flytende gasser eller damper (for eksempel vann, benzen, etc.) absorberes som et resultat av polymolekylær adsorpsjon. Hvori polymolekylært lag representerer tynn film av væske som dekker den indre overflaten av poren. Lag av en slik væske fusjonerer med hverandre på trange steder danner konkave menisker, under hvilke damptrykk dannes. Derved porene trekke inn gassmolekyler (damp) og fylt med væske dannet under kondensering.
Når det flyter adsorpsjon komplisert av kapillærkondensasjon, isotermen som tilsvarer fyllingen av porene (1) faller ikke sammen med isotermen (2) som tilsvarer deres tømming (fig. 18). På isotermen, kondenshystereseløkke. Prosessene for adsorpsjon og desorpsjon faller ikke sammen.
